 forcefield forcefield
forcefield forcefield Jump to Description or
Examples; explanation of how
commands are documented.
Jump to Description or
Examples; explanation of how
commands are documented.
Path/Keyword Values Default Meaning
select string $FORCEFIELD Name of forcefield contained in forcefield file.
How to handle missing parameters:
Nonbond parameters:
van der Waals parameters:
Coulomb parameters:
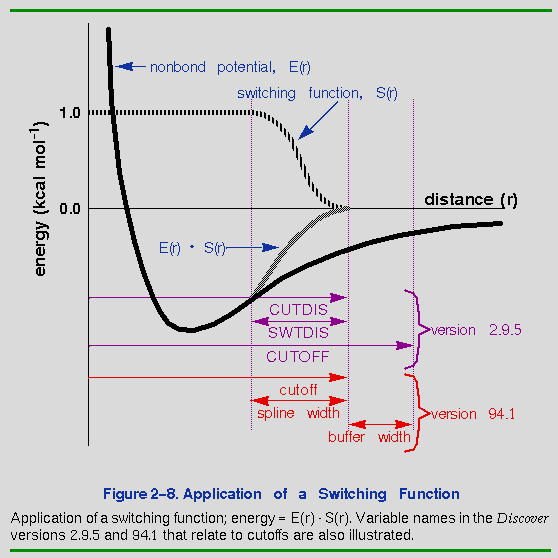
coulomb Pathname to Coulomb parameters. summa*tion_method group*_based group_based Method by which energy terms are atom*_based summed in the calculation when ewald generating the nonbond list. cell_m*ultiple (Note on cell multipole method.) cell_b*ased no_cutoff cutoff real 9.5 Distance (angstroms) at which to exclude interactions from the nonbond list for atom_based and group_based methods (used only if no_cutoff is not used). spline*_width real 1.0 Size (angstroms) of region within which nonbond interactions are splined from their full value to zero for atom_based and group_based methods (used only if no_cutoff is not used). buffer*_width real 0.5 Distance (angstroms) beyond cutoff at which nonbond interactions are zero (but the atoms are included in the nonbond list) for atom_based and group_based methods (used only if no_cutoff is not used). update*_width real 1.0 Distance (angstroms) an atom may move before nonbond list is regenerated, for ewald, cell_multipole, and cell_based methods (always applies to both van der Waals and Coulomb nonbond lists). (Important note) cmm_level second second Highest-order multipole and third highest-order Taylor series fourth expansion coefficient used in cell multipole nonbond calculation. number_of_cells integer 1 Number of layers of cells to include in nonbond list, counting from the base cell, for cell_multipole and cell_based summation methods (e.g., 1 means all the cells one unit cell away-- the 26 immediate neighbor cells). ewald_acc*uracy real 0.0025 Accuracy (kcal mol-1) used in Ewald calculations. ewald_di*pole Boolean false If true, then contributions from dipole moment are taken into account in Ewald calculations. distance_dep*endent_dielectric Boolean false Whether to use constant or distance-dependent dielectric for calculating Coulombic interactions. dielect*ric_value real 1.0 Magnitude of dielectric constant relative to
0.
Parameters to scale the energy terms:
scale Pathname to scale parameters. bond real 1.0 Scale the bond energy terms. angle real 1.0 Scale the angle energy terms. torsion real 1.0 Scale the torsion energy terms. oop real 1.0 Scale the out-of-plane energy terms. vdw real 1.0 Scale the van der Waals energy terms. coulomb real 1.0 Scale the Coulombic (charge) energy terms. cross real 1.0 Scale all the cross terms. hbond real 1.0 Scale hydrogen-bond energy terms. vdw_1_4 real 0.5 Scale the 1-4 van de Waals interactions for the Amber forcefield. No effect for other forcefields. coulomb_1_4 real 0.5 Scale the 1-4 Coulombic interactions for the Amber forcefield. No effect for other forcefields.
Jump to Syntax or
ExamplesThe forcefield command may be used to select one of the forcefields contained in a forcefield file. If this command is not specified, the default is to use the forcefield with the same name as the file. This command is currently available only with the CVFF forcefield file. This forcefield file contains the following forcefields:
The _stop parameters are used to control whether the Discover program exits with an error when parameters are not found for any interaction. The default behavior is that the Discover program stops if any bond, angle, torsion, or out-of-plane term is not found but continues for unfound cross terms, just setting the parameters for any missing cross terms to zero.
The forcefield command is also used to control the methods used in calculating nonbond interactions. Some of the methods available for nonbonds are:
The lists are automatically updated whenever any atom moves more than buffer_width/2. This prevents any discontinuities from arising due to atoms moving from outside the nonbond list directly into the spline region.
The forcefield scale command offers control over the individual types of energy terms in the forcefield.
Note that the forcefield command differs from other BTCL commands in that the parameter values specified are retained, i.e., the parameter default values are set when the command is performed. For example:
cdlConfig forcefield select = cvff_nocross parameters bond_automatic = FALSE scale angle = 1.1
forcefield scale bond = 1.1would be identical in functionality and operation to:
forcefield select = cvff_nocross parameters bond_automatic = FALSE scale angle = 1.1
forcefield scale bond = 1.1Whether to scale 1-4 interactions is determined by the keyword amber in the name of the forcefield file--if the word amber is not part of the name of the forcefield used, 1-4 scaling is not done. 1-4 van der Waals and 1-4 Coulombic interactions are scaled separately (as required by the new AMBER forcefield). The default scale factors are 0.5 for both. These two scale factors (vdw_1_4 and coulomb_1_4) just like any other scale factors. However, they have no effect on the energies if amber is not part of the name of the forcefield used.
Note on update_width keyword: For the Ewald and cell multipole methods, the update frequency of neighbor lists or Taylor coefficients is determined by update_width. If update_width is small (e.g. 0.0000001 Å), the neighbor lists or Taylor coefficients may be updated at every energy evaluation. By default (update_width = 1.0 Å), the neighbor lists or Taylor coefficients are updated whenever an atom moves more than update_width from its original position. Thus, the number of steps of dynamics or minimization that occur between updates increases. In minimization runs, an energy discontinuity may result. This is because the neighbor lists or Taylor coefficients are created at one geometry and used for energy evaluations at that geometry as well as the slightly different geometries of subsequent steps. The discontinuity becomes apparent if the final geometry is evaluated later (in a subsequent energy command), since new neighbor lists and Taylor coefficients are generated. An appropriate value of update_width depends on the application. More frequent updates of the Taylor coefficients slow down the cell multipole method, but make it more accurate. The discontinuity is insignificant for the Ewald method.
Note on Ewald accuracy: Although the default Ewald accuracy is acceptable for most single-point energy calculations, a tighter accuracy may be required for some minimization and dynamics runs, to insure acceptable gradient accuracy. In particular, using 0.00025 may be preferable if your minimization run fails to converge or your dynamics run misbehaves.
Note on cell multipole method: Currently, the cell multipole method does not calculate second derivatives. For nonperiodic systems with fewer than 200 atoms, the cell multipole method is switched to the no_cutoff method if second derivatives are requested.
Also note that the cell_multipole method does not calculate virials (cell derivatives). Thus, cell minimization and constant-pressure dynamics cannot be carried out using this nonbond method.
If use_quartic is set to true, the cutoff parameter is not used. The nonbond repulsive energy is calculated using the quartic potential formula and is included only when the distance between the pair of atoms is less than the scaled van de Waals radius. The nonbond dispersive and Coulombic parts are automatically turned off. Also the atom_based nonbond summation method is automatically used. The use_quartic keyword should be set to false explicitly if a normal calculation is desired after the use of the quartic potential.
The Buckingham (exponential-6) nonbond interaction potential is also supported. To use this potential form, a section containing the appropriate forcefield parameters should be inserted into the forcefield file, for example:
#nonbond(exp-6) cvff > E = Aij * exp(-r/rho) - Cij/r^6 !Ver Ref I J A rho C !---- --- ---- ------- -------- --------- -------- 1.0 14 LR O2 6740.43800 0.34720 0.00000 1.0 14 LS OS 6740.43800 0.34720 0.00000
Jump to Syntax or
Description
forcefield select = cvff_nocrossThis example of the forcefield command selects the cvff_nocross forcefield from the current forcefield file. Use of this command causes the energy expression to be recreated with the new forcefield parameters the next time an energy evaluation is performed.
forcefield parameters bond_automatic = FALSE \ angle_automatic = FALSE \ torsion_automatic = FALSE \ oop_automatic = FALSEThis example of the forcefield command disables all use of automatic parameters.
forcefield nonbond vdw summation_method = atom_basedThis example of the forcefield command specifies that subsequent calculations use the atom-based method to generate the nonbond summation list. All other forcefield parameters are set to their current defaults.
forcefield scale angle = 1.5This example of the forcefield command indicates that the contributions of the angle energy terms are to be multiplied by 1.5. All other forcefield parameters are set to their current defaults.
forcefield nonbond +separate_coulomb_cutoffs \ coulomb cutoff = 12 spline_width = 1.5 \ buffer_width = 0.5In this example, the defaults specify the use of separate nonbond lists for the van der Waals and Coulombic energy terms and set the Coulomb list cutoff to 12 Å, the Coulomb spline width to 1.5 Å, and the Coulomb buffer width to 0.5 Å. This corresponds to a spline region going from the full value at 10.0 Å down to zero at 11.5 Å (followed by a 0.5-Å buffer region before the cutoff at 12.0 Å).
forcefield scale \
vdw_1_4 = 0.4 \
coulomb_1_4 = 0.4
This BTCL statement is used to scale both 1-4 van der Waals and 1-4
Coulombic interactions by a factor of 0.4 for the AMBER
forcefield. The statement has no effect if the forcefield used is not
AMBER.
 Main
access page
Main
access page  Advanced-Use access.
Advanced-Use access.
 List of BTCL commands
List of BTCL commands
 energyContribution command
energyContribution command
 geometry command
geometry command
Copyright Biosym/MSI
{kind=link}