 Assignment of Forcefield
Atom Types Assignment of Forcefield
Atom Types
Assignment of Forcefield
Atom Types Assignment of Forcefield
Atom TypesAs a more complicated example, consider propane, which has four distinct types of atoms: methyl carbon atoms, methyl hydrogen atoms, a methylene carbon atom, and the methylene hydrogens. In principle, a forcefield could consider these to be four distinct atom types, but in practice, the chemical difference between the carbon atoms or between the hydrogen atoms is very small, so in most forcefields the carbon atoms are all assigned the same atom type, and all the hydrogens are assigned a second atom type.
Atom types must be assigned externally for the Discover program, and it reads the atom type information from the molecular data file (.mdf). The molecular data file and Cartesian coordinate file (.car) are often created in the Insight molecular modeling program, which takes care of assigning the atom types. The Discover program also expects to read partial atomic charges from the .mdf file, because the Discover program has no mechanism for determining these charges. Again, the Insight program is usually used to assign the charges.
It is important to thoroughly understand how the Discover program associates the parameters from the forcefield with individual internal coordinates, because the energy, derivatives, structures, and almost all other properties calculated by the Discover program depend on these forcefield parameters and the way in which they are associated with the internal coordinates. The following sections describe three facets of this process: atom type equivalences, wildcards in parameter definitions, and automatic parameter assignments.
The actual format of the equivalence data in the forcefield parameter file is detailed in the printed Files book. For the equivalences used in any particular forcefield, you should examine the actual forcefield parameter file for up-to-date information.
Missing parameters in the CFF91 and CVFF forcefields are automatically assigned by switching to an automatic forcefield. This switching is accomplished with an equivalence table that converts the original set of atom types to a smaller set of generic atom types.
In the automatic forcefield, the atom types for bonds, angles, torsions, and out-of-plane deformations have different levels of specificity. For example, while bond-stretching parameters are determined by the atom types of both atoms; angle-bending and torsion parameters may be determined by the atom type of only the central atom(s). A wildcard (*), representing any type of atom, is used for the end atoms of torsions and angles.
In some cases, angle-bending parameters are specified by two atoms (rather than only the central atom). This can lead to ambiguity-for example, C-C-N can be associated with c_-c_-* or with n_-c_-*. The underscore in this example is used to denote the generic (or automatic) atom types. Here, a one-sided wildcard (*#, where # is an integer indicating the precedence), is used for one of the end atoms in an angle. The parameters for a C-C-N angle would be taken from those for atom types n_-c_-*6 rather than c_-c_-*7, because 6 is smaller than 7.
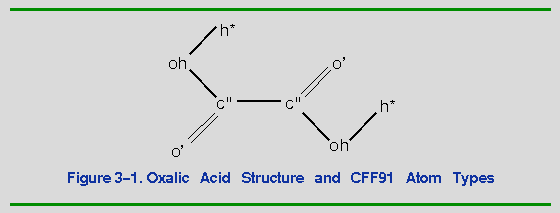
As an example, the parameters for the angle oh-c"-c" in oxalic acid (Figure 3-1) are not present in CFF91.
When the automatic parameter assignment process is used, it looks at the auto-equivalence table in the cff91.frc file to find the generic atom types for this angle (indicated in bold type):
#auto_equivalence cff91_auto ! Equivalences ! ----------------------------------------------------------------------------- !Ver Ref Type NonB Bond Bond Angle Angle Torsion Torsion OOP OOP ! Inct End Atom Apex Atom End Atoms Center Atoms End Atom Center Atom !--- --- ---- ---- ---- --- -------- ---------- ---------- ------------ -------- ----------- 2.0 2 c" c" c" c'_ c_ c'_ c_ c'_ c_ c'_ 2.0 2 oh o o_ o_ o_ o_ o_ o_ o_ o_Thus, atom type oh is reassigned to o_, c" is reassigned to c'_ for the apex atom, and c" is reassigned to c_ for the end atom. The parameters for the oh-c"-c" angle are taken from the o_ c'_ *7 line in the quadratic_angle section of the cff91.frc file:
#quadratic_angle cff91_auto > E = K2 8 (Theta - Theta0)^2 !Ver Ref I J K Theta0 K2 !---- --- ---- ---- ---- --------- --------- 2.0 2 c_ c'_ *9 120.0000 40.0000 2.0 2 n_ c'_ *8 120.0000 53.5000 2.0 2 o_ c'_ *7 110.0000 122.0000 2.0 2 o'_ c'_ *f 120.0000 68.0000 2.0 2 h_ c'_ *2 110.0000 55.0000
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Forcefields access
Forcefields access
 Forcefields Supported by Discover Program
Forcefields Supported by Discover Program
 CVFF Forcefield
CVFF Forcefield
Copyright Biosym/MSI
{kind=link}