 Cell Multipole Method Cell Multipole Method
Cell Multipole Method Cell Multipole Method
The cell multipole method applies to the general energy expression of the following form:
The general potential i
may be divided into a near-field potential due to the surrounding
atoms (those within a few angstroms) and a far-field potential due to
the rest of the atoms that interact with the ith atom.
The number of interactions in the near field is limited, so it is relatively easy to calculate the near-field potential exactly. The number of interactions in the far field is of order N 2, making an exact calculation of this potential intractable for large molecules. The cell multipole method calculates the far-field potential accurately and efficiently as described under Implementation of the CMM Method.
The cell multipole method involves the following two key steps:
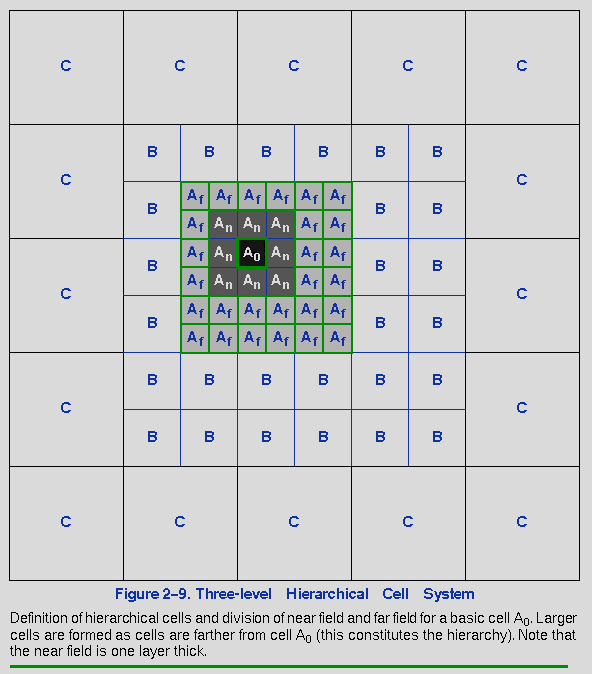
The potential associated with each basic cell can be represented as a general potential originating at the center of the cell. This potential may be expanded into an infinite series of multipole moments. For example, the potential associated with cell Af in Figure 2-9 centered at rAf, is expressed as:
where R = rAf - r; r is any point outside cell Af;
The potentials associated with the higher-level cells can be expanded in an analogous manner, with moments derived from lower-level cell moments.
Using this expansion to represent the potentials associated with Af-, B-, and C-level cells, the far-field potential of cell A0 may be obtained by summing all the far-cell contributions. The resulting potential may now be expanded as a Taylor series about the center of cell A0:
where rA0 is the position vector of the center of cell A0 and
A key point of the cell multipole method is that, once the set of Taylor coefficients is calculated at rA0, the far-field potential of any atom in cell A0 is obtained easily through Eq. 2-9.
Since the Taylor coefficients must be generated for every basic cell, another key point of the cell multipole method is efficient generation of these coefficients. A hierarchical procedure is used, in which coefficients determined for higher-level cells are propagated to the coefficients for lower-level cells. Thus, coefficients for a child B cell are obtained by adding contributions directly translated from the C-level coefficients at the center of the parent C cell to the coefficients at the center of B, generated by considering only the B-cell contributions.
For larger systems than hemoglobin, the improvement in performance can be even more dramatic. For a system ten times larger, the cell multipole method would take 3-10 minutes for the energy evaluation, depending on the accuracy desired. The exact N 2 calculation, in contrast, would take about three days!
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Describe-System access
Describe-System access
 Nonbond Cutoffs
Nonbond Cutoffs
 Ewald Sums
Ewald Sums
Copyright Biosym/MSI
{kind=link}