 Consistent Valence
Forcefield (CVFF)
Consistent Valence
Forcefield (CVFF)
Contents
The consistent-valence forcefield (CVFF), the original forcefield
provided with the Discover program, is a generalized valence
forcefield (Dauber-Osguthorpe
1988). Parameters are provided for amino acids, water, and a
variety of other functional groups.
The new version of the Discover program (95.0/300) has
undergone extensive validation tests comparing it with the previous
version of Discover (2.x.x). These tests have indicated that
the two programs provide exactly the same results for all components
of the energy expression with one exception: the out-of-plane energy
for the CVFF forcefield.
The out-of-plane energy for the CVFF forcefield is calculated as an
improper torsion. Recall that an improper torsion views three
connected atoms and a central atom as a torsion (e.g., if A2 is the
central atom, construct a torsion as A1-A2-A3-A4--this last connection
does not represent a real bond, hence the name improper
torsion). There are three possible improper torsions that can be
generated for a particular out-of-plane based on permutations of the
connected atoms.
For CVFF, only one of these improper torsions is used. The rules that
Discover 2.x.x employs to select the particular improper
torsion are somewhat arbitrary, and it is not possible to replicate
them in the Discover 95.0/300 program. However, the changes in
energy are very small (on the order 0.01 kcal mol-1). A
more rigorously defined out-of-plane, the Wilson out-of-plane, is used
in the CFF forcefield. This energy term
provides exact agreement between the two programs.
The analytic form of the energy expression used in CVFF is given in Eq. 3-1. Most other forcefields in the literature
use a subset of the terms included in CVFF, often only the diagonal
terms.
- Eq. 3-1:
-
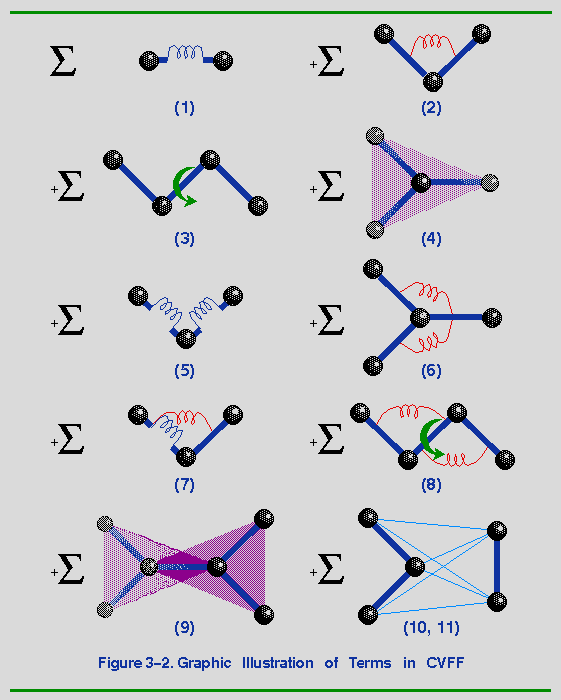
Eq. 3-1 is illustrated schematically in Figure 3-2. Terms 1-4 in Figure
3-2 and Eq. 3-1 are commonly referred to as the diagonal terms of the
valence forcefield and represent the energy of deformation of bond
lengths, bond angles, torsion angles, and out-of-plane interactions,
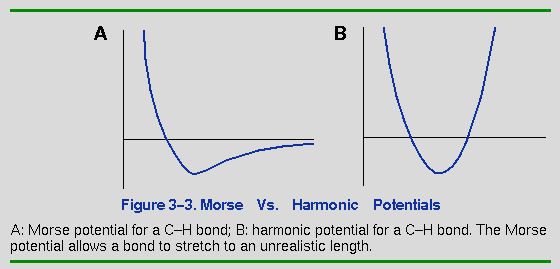
respectively. Note that a Morse potential (Term 1) is used for the
bond-stretching term. The Discover program also supports a
simple harmonic potential for this term. The Morse form is
computationally more expensive than the harmonic form. Since the
number of bond interactions is usually negligible relative to the
number of nonbond interactions, the additional cost of using the more
accurate Morse potential is insignificant, so this is the default
option.
One exception to this rule is when the molecule being simulated is
high in energy (caused, for example, by overlapping atoms or a high
target temperature), which might force a Morse-style function to allow
the bonded atoms to drift unrealistically far apart (see Figure 3-3).
Terms 5-9 in Figure 3-2 and
Eq. 3-1 are off-diagonal (or cross) terms and represent couplings
between deformations of internal coordinates. For example, Term 5
describes the coupling between stretching of adjacent bonds. These
terms are required to accurately reproduce experimental vibrational
frequencies and, therefore, the dynamic properties of molecules. In
some cases, research has also shown them to be important in accounting
for structural deformations. However, cross terms can become unstable
when the structure is far from a minimum. Therefore, although the
Discover program includes cross terms by default, input files
created by the Insight program by default explicitly turn off
the cross terms.
Terms 10-11 describe the nonbond interactions. Term 10 represents the
van der Waals interactions with a Lennard-Jones function. Term 11 is
the Coulombic representation of electrostatic interactions. The
dielectric constant  can be made
distance dependent (i.e., a function of
rij ). In the CVFF forcefield, hydrogen bonds
are a natural consequence of the standard van der Waals and
electrostatic parameters, and special hydrogen bond functions do not
improve the fit of CVFF to experimental data (Hagler 1979a, 1979b).
can be made
distance dependent (i.e., a function of
rij ). In the CVFF forcefield, hydrogen bonds
are a natural consequence of the standard van der Waals and
electrostatic parameters, and special hydrogen bond functions do not
improve the fit of CVFF to experimental data (Hagler 1979a, 1979b).
Additional information on the forcefields and how they can be
augmented is contained in the printed Files book, where the .frc file
is described.
The CVFF forcefield supplied by Biosym/MSI defines atom types for the 20
commonly occurring amino acids, most hydrocarbons, and many other
organic molecules (Table 3-1).
The bond increment sections of the .frc files for both CFF91 and CVFF
have been expanded so that partial charges can be determined whenever
the Discover program is able to assign automatic atom types.
The format is:
- atom type
- description
and you may quickly jump to the classes of atom types by clicking:
- d
- general deuterium (equiv. to h)
- dw
- deuterium in heavy water (equiv. to h*)
- h
- generic hydrogen bonded to C, Si, or H
- hc
- hydrogen bonded to C (equiv. to h)
- hi
- hydrogen in charged imidazole ring (equiv. to hn)
- hn
- hydrogen bonded to N
- ho
- hydrogen bonded to O
- hp
- hydrogen bonded to P (equiv. to h)
- hs
- hydrogen bonded to S
- hw
- hydrogen in water (equiv. to h*)
- h*
- hydrogen in water
- h+
- charged hydrogen in cation (equiv. to hn)
- c
- generic sp3 carbon
- ca
- general amino acid alpha carbon (sp3)
(equiv. to cg)
- cg
- sp3 alpha carbon in glycine
- ci
- sp2 aromatic carbon in charged
imidazole ring (his+ )
- cn
- sp3 carbon bonded to N (equiv. to cg)
- co
- sp3 carbon in acetal (equiv. to c)
- coh
- sp3 carbon in acetal with hydrogen
(equiv. to cg)
- cp
- sp2 aromatic carbon (partial double
bonds)
- cr
- carbon in guanidinium group (HN=C(NH2)2)
(arg)
- cs
- sp2 carbon in 5-membered ring next to S
- ct
- sp carbon involved in triple bond
- c1
- sp3 carbon bonded to 1 H, 3 heavy
atoms (equiv. to cg)
- c2
- sp3 carbon bonded to 2 H's, 2 heavy
atoms (equiv. to cg)
- c3
- sp3 carbon in methyl (CH3) group
(equiv. to cg)
- c5
- sp2 aromatic carbon in 5-membered
ring
- c3h
- sp3 carbon in 3-membered ring with
hydrogens (equiv. to cg)
- c3m
- sp3 carbon in 3-membered ring
(equiv. to c)
- c4h
- sp3 carbon in 4-membered ring with
hydrogens (equiv. to cg)
- c4m
- sp3 carbon in 4-membered ring
(equiv. to c)
- c
- sp2 carbon in carbonyl (C=O) group of
amide
- c"
- carbon in carbonyl group, not amide (equiv. to c)
- c*
- carbon in carbonyl group, not amide (equiv. to c)
- c-
- carbon in charged carboxylate (COO- ) group (equiv. to c)
- c+
- carbon in guanidinium group (equiv. to cr)
- c=
- nonaromatic end doubly bonded carbon
- c=1
- nonaromatic, next-to-end doubly bonded carbon
- c=2
- nonaromatic doubly bonded carbon
- n
- generic sp2 nitrogen in amide
- na
- sp3 nitrogen in amine (equiv. to n3)
- nb
- sp2 nitrogen in aromatic amine
(equiv. to n3)
- nh
- sp2 nitrogen in 5- or 6-membered
ring, with hydrogen attached (equiv. to np)
- nho
- sp2 nitrogen in 6-membered ring, next
to a carbonyl group and with a hydrogen (equiv. to np)
- nh+
- protonated nitrogen in 6-membered ring
- ni
- sp2 nitrogen in charged imidazole
ring (his+)
- nn
- sp2 nitrogen in aromatic amine
(equiv. to n3)
- np
- sp2 nitrogen in 5- or 6-membered ring
- npc
- sp2 nitrogen in 5- or 6-membered
ring, bonded to a heavy atom (equiv. to np)
- nr
- sp2 nitrogen in guanidinium group
(HN=C(NH2)2)
- nt
- sp nitrogen involved in triple bond
- nz
- sp nitrogen in N2
- n1
- sp2 nitrogen in charged arginine
- n2
- sp2 nitrogen in guanidinium group
(HN=C(NH2)2)
- n3
- sp3 nitrogen with 3 substituents
- n4
- sp3 nitrogen in protonated amine
- n3m
- sp3 nitrogen in 3-membered ring
(equiv. to n3)
- n3n
- sp2 nitrogen in 3-membered ring
(equiv. to n)
- n4m
- sp3 nitrogen in 4-membered ring
(equiv. to n3)
- n4n
- sp2 nitrogen in 4-membered ring
(equiv. to n)
- n+
- sp3 nitrogen in protonated amine
(equiv. to n4)
- n=
- nonaromatic end doubly bonded nitrogen
- n=1
- nonaromatic, next-to-end doubly bonded nitrogen
- n=2
- nonaromatic doubly bonded nitrogen
- o
- generic sp3 oxygen
- oc
- sp3 oxygen in ether or acetal
(equiv. to o)
- oe
- sp3 oxygen in ester (equiv. to o)
- oh
- oxygen bonded to H
- op
- sp2 aromatic oxygen in 5-membered ring
- o3e
- sp3 oxygen in 3-membered ring
(equiv. to o)
- o4e
- sp3 oxygen in 4-membered ring
(equiv. to o)
- o
- oxygen in carbonyl (C=O) group
- o*
- oxygen in water molecule
- o-
- oxygen in charged carboxylate (COO- ) group
- s
- sp3 sulfur
- sc
- sp3 sulfur in methionine (C-S-C)
group (equiv. to s)
- sh
- sulfur in sulfhydryl (SH) group
- sp
- sulfur in aromatic ring, e.g., thiophene
- s1
- sulfur involved in S-S disulfide bond (equiv. to s)
- s3e
- sulfur in 3-membered ring (equiv. to s)
- s4e
- sulfur in 4-membered ring (equiv. to s)
- s
- sulfur in thioketone (>C=S) group
- s-
- partial-double sulfur bonded to something that is bonded to another
partial-double oxygen or sulfur
- p
- general phosphorous atom
- br
- bromine bonded to a carbon
- cl
- chlorine bonded to a carbon
- f
- fluorine bonded to a carbon
- i
- covalently bound iodine
- Br
- bromide ion
- ca+
- calcium ion (Ca2+)
- Cl
- chloride ion
- Na
- sodium ion
- ar
- argon atom
- si
- silicon atom
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Forcefields access
Forcefields access
 Atom Type Assignment
Atom Type Assignment
 CFF91 Forcefield
CFF91 Forcefield
Copyright Biosym/MSI
{kind=link}
{kind=link}