 Nonbond Cutoffs Nonbond Cutoffs
Nonbond Cutoffs Nonbond Cutoffs
To appreciate the impact of cutoffs on computational efficiency, consider a protein-substrate-solvent system with 5000 total atoms. A system of this size would be typical of a small protein (100-150 residues) surrounded by 1-2 layers of water.
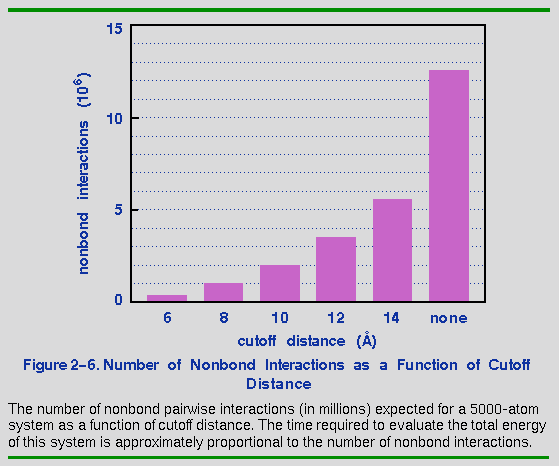
Figure 2-6 shows how the number of nonbond interactions increases with the cutoff distance. This calculation would run at least 10 times faster with an 8.0 Å cutoff than with no cutoff (assuming that the nonbond term is rate limiting, which it usually is). The trade-off is, of course, that interactions beyond the cutoff distance are not accounted for. Both van der Waals interactions and electrostatic interactions are significant up to 15 Å or more. For example, in a recent calculation of the energy as a function of cutoff distance in the [Ala-Pro-D-Phe]2 crystal, Kitson and Hagler showed that the nonbond energy accounted for changes from 63 to 97% of the asymptotic value as the cutoff distance was increased from 8 to 15 Å (Kitson and Hagler 1988).
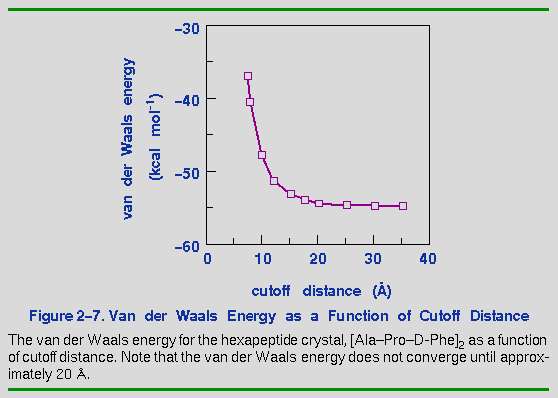
Figure 2-7 shows how the van der Waals component of the nonbond energy varies as a function of cutoff distance for the [Ala-Pro-D-Phe]2 crystal. The van der Waals energy changes by 40% when the cutoff distance is increased from 8 to 15 Å. The exact dependence of the energy on the cutoff distance depends on the system itself and should be calibrated for each new system.
For efficiency, the Discover program creates a neighbor list containing all pairs of atoms to be used during calculation of the nonbond interactions. Since this list is not updated at every step of the calculation, it includes atoms in a buffer region that might move close enough to contribute before the next update of the neighbor list. In the Discover 2.9 program the buffer region is defined by the difference between CUTDIS and CUTOFF, and in the Discover 95.0 program the buffer width is explicitly specified, along with the cutoff. To ensure that no atoms outside the buffer region can move close enough to interact, the list is automatically updated whenever any atom moves more than one-half the buffer width. Thus, the width of this buffer region, coupled with the velocity with which atoms move, determine the maximum time before the neighbor list is updated.
To understand the implications, note that the interaction of two monopoles, each of one electronic unit of charge, is about 33 kcal mol-1 at 10 Å, while the interaction energy of two dipoles formed from unit monopoles is no more than about 0.3 kcal mol-1. It is clear that ignoring the monopole-monopole interaction would be grossly misleading, whereas ignoring the dipole-dipole interaction would only be a modest approximation. If the nonbond cutoffs were applied on an atom-by-atom basis, they could artificially split dipoles by having one atom inside the cutoff and one atom outside. Instead of ignoring a relatively small dipole-dipole interaction, this would have the effect of artificially introducing a large, monopole-monopole interaction. To avoid these artifacts, the Discover program applies cutoffs over charge groups.
A charge group is a small group of atoms close to each other which have a net charge of zero or almost zero. In many cases, charge groups are identical to common chemical functional groups. Thus, a carbonyl group, methyl group, or carboxylic group would also be an approximately neutral charge group. The Discover program designates one atom from each charge group as the switching atom. The Discover program generates the neighbor list by considering the distance between the switching atoms of two groups. If the distance is less than the cutoff distance, then all the atom pairs in the two groups are included. If the distance is greater than the cutoff, they are all excluded. Similarly, when calculating the actual interaction energy, the Discover program switches off the interaction between all atom-atom pairs in the two groups based only on the distance between the two switching atoms. This procedure prevents artifacts due to splitting of dipoles.
The size of a charge group, as defined by the greatest distance from the switching atom to another atom in the group, must be significantly smaller than the cutoff distances. Otherwise, the interaction between two atoms close to each other might be ignored because the switching atoms of the two groups are farther apart than the cutoffs. Typical groups are no more than 1-3 Å large, so cutoffs larger than 7-8 Å are reasonable. However, occasionally molecules contain considerably larger groups. The Discover program checks the size of the groups against the cutoff distances, outputs an error message, and terminates if it decides the cutoffs are too short compared to the group size. In this case, you must either increase the cutoffs or redefine the groups to be smaller. The Discover program also warns you about significantly non-neutral groups. Some of these can be expected if the molecule contains formally charged functional groups, such as protonated amines and carboxylates. However, other non-neutral groups usually indicate an error in group definitions.
The Discover program also incorporates an improvement over a single cutoff distance called double cutoffs, or, as it is sometimes called in dynamics, multiple timesteps. The nonbond interaction potential at a distance is a smooth function that does not vary rapidly. With double cutoffs, two cutoff distances--an inner and outer one--are assigned. The two distances define an inner spherical region and an outer shell around a given atom. Interactions with atoms in the inner region are treated as usual, but interactions with the atoms in the outer shell are updated only at every neighbor list update. It is assumed that their contribution varies only slowly. These double cutoffs are used to make the calculation less expensive by reducing the inner cutoff to a smaller value than could normally be used with a single cutoff. Accuracy is regained at minimal cost by using a large distance for the outer cutoff.
It is important to realize that the effective potential energy surface is not quite continuous when double cutoffs are used. The magnitude of the discontinuities depends on the cutoff distances and the system that is being studied. These discontinuities are only a minor problem for dynamics, where they are manifested as small fluctuations in the total energy. Their effect during minimization depends on the minimizer that is used, because some minimization algorithms, such as conjugate gradients, are quite sensitive to discontinuities in the surface. Other algorithms, such as steepest descents, are relatively robust.
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Describe-System access
Describe-System access
 Periodic Boundary Conditions
Periodic Boundary Conditions
 Cell Multipole Method
Cell Multipole Method
Copyright Biosym/MSI
{kind=link}
{kind=link}