 Dynamics - General
Methodology Dynamics - General
Methodology
Dynamics - General
Methodology Dynamics - General
Methodology
Studies of molecular motions can be used to derive properties such as diffusion coefficients.
Real molecules find their conformational global minimum by fluctuating about an ensemble of configurations within energetic reach (as determined by the system temperature). In principle then, if the motions and fluctuations experienced by a molecule are simulated, the global minimum is sampled by this simulation (eventually). This is the approach taken by molecular dynamics. In molecular dynamics, the fact that the negative of the gradient of the potential energy is equivalent to the force is used. Given the force and mass for each atom, Newton's equation of motion (F = ma) can be numerically integrated to predict where atoms will move within a short time interval. By using successive time steps, a time-dependent trajectory can be constructed for all atoms which thus represents the molecular motions.
By using the available thermal energy to climb and cross conformational energy barriers, dynamics provides insight into the accessible conformational states of the molecule. Molecular dynamics has been used in a variety of small peptide systems to find lower-energy conformational states across energy barriers that would be inaccessible to classical minimization. In particular, it has been applied to the study of vasopressin (Hagler 1985) and gonadotropin-releasing hormone (GnRH) (Struthers et al. 1984, Hagler 1985).
For GnRH, novel constrained analogs with 100-fold higher potency than the parent cyclic compounds were designed, based on the conformations of GnRH and GnRH analogs identified with a combined strategy incorporating energy minimization, template forcing, and molecular dynamics.
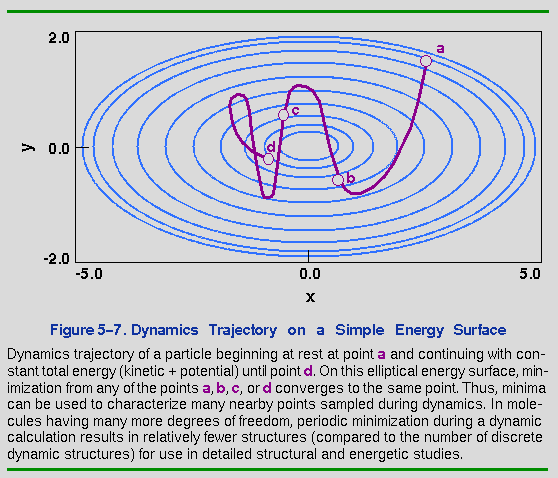
One of the disadvantages of dynamics, however, is that many hundreds and even thousands of conformational states are sampled during a trajectory, thus compounding the analysis process. Furthermore, the transitions of interest may take place on a time scale of milliseconds to minutes or longer at 300° and, at present, dynamic simulations are limited to tens of nanoseconds. Minimizing periodically during a dynamics trajectory helps the analysis problem by identifying those structures that represent the local minimum about which the dynamics fluctuations take place. This is illustrated in Figure 5-7, where the dark magenta line superimposed on a simple elliptical energy surface represents a dynamics trajectory of a particle beginning at rest from point a. The initial gradient pulls the particle downhill but, because energy is conserved, its velocity increases as it gets closer to the minimum. Its momentum takes the particle back and forth, visiting many points on the energy surface during the trajectory. However, if we stop the particle at any point during the dynamics and minimize, only one point is found. In higher-dimensional systems, such as real molecules, minimization from various dynamics points may result in the identification of several different local minima. However, the number of minima is far fewer than the number of points sampled during dynamics, and these minima are useful as points of reference in analyzing molecular structures and energetics.
Of course, a real molecular system has far too many degrees of freedom to allow plotting the trajectory on a realistic energy surface. Any attempt to project a trajectory into one or two dimensions results in a far more complex pattern that obscures which points, if any, are close to a minimum. In real calculations, then, periodic (systematic) minimizations must be used to discover which minima are being sampled by dynamics. A detailed discussion of how dynamic searching has been used to characterize the conformational states of the peptide hormone, atrial natriuretic factor (ANF) is presented as an illustration.
The strategy included these steps:
Mackay et al. (1989) performed a set of molecular dynamics calculations on ANF at elevated temperature to search for structural features that may be significant for ANF binding to its receptor. The goals of these calculations were: (1) to search the conformational space of the molecule; (2) to find unique regions that may be cross-linked to test conformational hypotheses; and (3) to compare different approaches using dynamics to search conformational space.
5 10
SER-LEU-ARG-ARG-SER-SER-CYS-PHE-GLY-GLY-ARG-MET-
/ ASP-
S ARG-
S ILE- 15
/ GLY-
TYR-ARG-PHE-SER-ASN-CYS-GLY-LEU-GLY-SER-GLN-ALA-
25 20
An initial geometry was generated by first building the sequence into
an extended conformation using standard amino acid bond lengths,
angles, and side chain dihedral angles. The disulfide bridge was
formed by imposing a Solvent was not included in this system, because the interest was not in the conformation of ANF in solution, but rather in ANF bound to its receptor. There was no explicit receptor model with which to interact; the objective was to look for any conformations accessible to the ANF regardless of the environment. High-energy structures were not ignored because the receptor may stabilize them, and the structures found were compared with those of other analogs. Common structural features among ANF and ANF analogs may suggest motifs that can be used to construct a putative active conformation.
Since solvent was not included, all the amino acids were defined to be in their neutral states, to prevent unscreened interactions between charged side chains from dominating the structural search. Strong salt bridges often form that constrain the molecule from finding alternative conformations during dynamics. This structure was then equilibrated by running dynamics at 300 K and 600 K for 20 ps each. Data were collected from a subsequent 100 ps run at 900 K. A total of 100,000 configurations (one every 10-15 seconds) was sampled during this simulation. To reduce the volume of data to a more manageable level, instantaneous dynamic structures were minimized at one-picosecond intervals, reducing the number of structures to be analyzed to 100.
High temperatures greatly increase the efficiency of producing conformational transitions. Consider a simple Arrhenius model for reaction rates:
The conformational search of ANF at 900 K produced at least 11 distinct families in 100 ps. A similar control run at 300 K produced no significant new conformational states over the same period. Dynamics at 900 K was not without risk, however. For example, the peptide bond was observed to have undergone trans-cis conversions in 42 of 100 minimized structures. This is clearly a result of the high temperature--no trans-cis transitions were observed in room temperature simulations. To avoid formation of cis bonds, a torsional restraint of 5 kcal mol-1 rad-2 was added to the peptide bond, which effectively eliminated trans-cis conversions. This is another good example of how restraints can be incorporated into modeling strategies. Without a restraint to prevent trans-cis interconversion, high-temperature dynamics would not be as useful for conformational searching of peptides.
Overall, minimization and structural classification strategies were used to refine 100,000 dynamic structures to 11 representative structures. This more manageable set of conformations was then analyzed in detail for important structural features to be incorporated into a strategy for designing analogs.
-C
distances was calculated for each of the
eleven representative annealed structures. These unique distances
might be used as the basis for such a strategy (Rodgers, to be
submitted).
The strategy includes these steps:
Thus, its structure includes a constrained ring of six residues and a flexible three-residue tail. Hagler et al. (1985) reported a dynamics study of vasopressin with the objective of characterizing the dynamic transitions and conformational equilibria of this flexible peptide.
As in the ANF example, minimization techniques play a major role in understanding and analyzing dynamic results.
-helix before eventually finding a
C7ax state, all in just over 6 ps. As was done
for ANF, structures found during the dynamics run were minimized to
confirm that the dynamics trajectory was passing through legitimate
minima and not just fluctuating about a single minimum. The
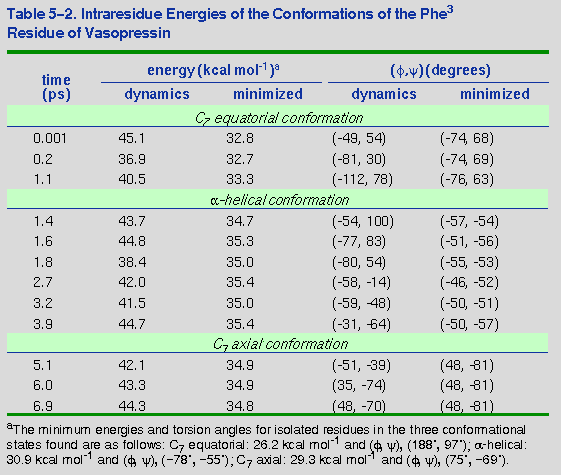
instantaneous conformational states, along with the resulting
minimized states, are shown in Table 5-2 for
Phe3. Note that dynamic structures as characterized by the
(
,
) values for
Phe3 can vary widely (by as much as 100 degrees) but still
minimize to the same conformational state. Nevertheless, there are
three distinct states to which the dynamic structures minimize. This
rigorously demonstrates that transitions are occurring.
One of the most significant results of this study was that the energy
levels for conformational states of Phe3 in vasopressin are
more closely spaced than for isolated phenylalanine. For example, the
difference between C7eq and the -helical state in an isolated blocked Phe
residue is calculated to be 4.7 kcal mol-1, while in
vasopressin it is only 2.2 kcal mol-1. This effective
leveling of the energy levels imposed by constraints in the ring
contributes to an enhanced flexibility in this part of the vasopressin
molecule.
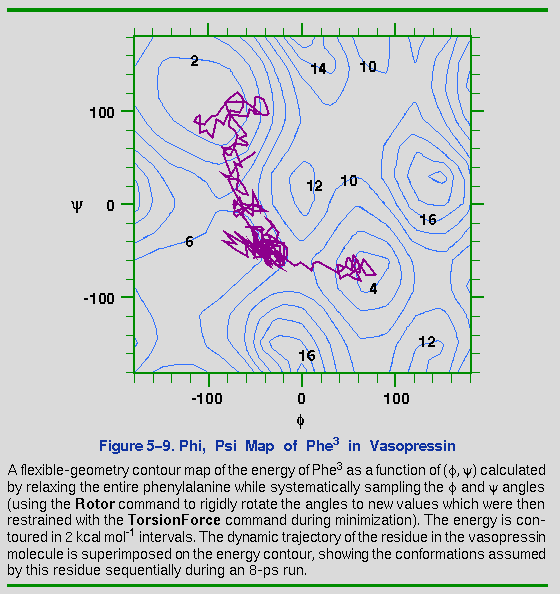
To illustrate this point, consider Figure 5-9, which superimposes
the Phe3 (,
) trajectory onto an adiabatic (that is,
flexible-geometry) contour surface. Note that the barrier,
particularly from the
-helix to the
C7ax conformations, is only 3-4 kcal
mol-1. The flexible-geometry torsion map is particularly
useful for understanding a dynamics trajectory, because it can nicely
explain the path the molecule follows on the energy surface.
The Constraints and
Restraints section includes torsion restraints, which are used to
prepare adiabatic torsional potential energy surfaces such as the ,
map used in Figure 5-9.
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Dynamics access
Dynamics access
 Dynamics - Constraints
Dynamics - Constraints
 References
References
Copyright Biosym/MSI
{kind=link}
{kind=link}
{kind=link}