 Minimization Algorithms Minimization Algorithms
Minimization Algorithms Minimization Algorithms
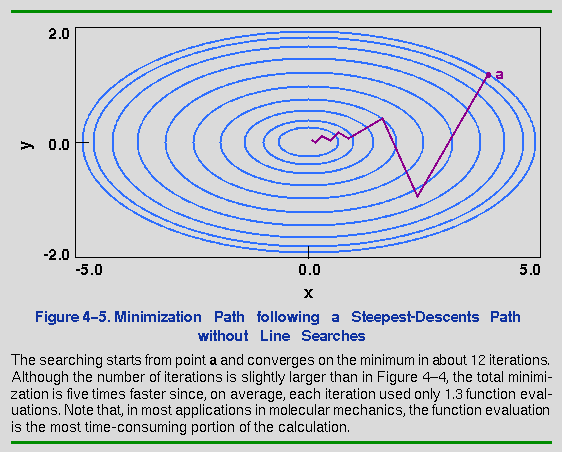
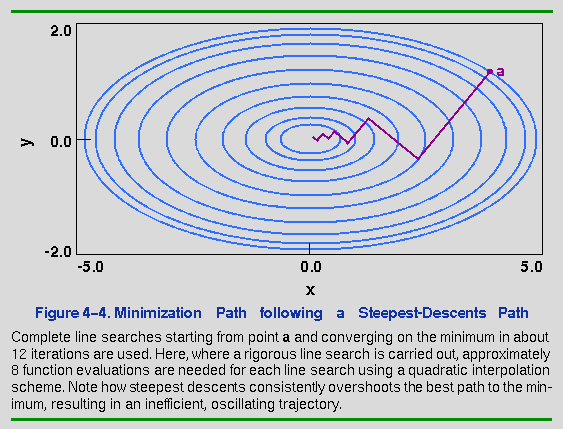
What would happen if the line search were eliminated and the position would simply be updated any time that the trial point along the gradient had a lower energy? The advantage would be that the number of function evaluations performed per iteration would be dramatically decreased. Furthermore, by constantly changing the direction to match the current gradient, oscillations along the minimization path might be damped. The result of such a minimization is shown in Figure 4-5. The minimization begins from the same point as in Figure 4-4, but each line search uses, at most, two function evaluations (if the trial point has a higher energy, the step size is adjusted downward and a new trial point is generated). Note that the steps are more erratic here, but the minimum is reached in roughly the same number of iterations. The critical aspect, however, is that by avoiding comprehensive line searches, the total number of function evaluations is only 10-20% of that used by the rigorous line search method. The Discover program uses the latter method for its steepest-descents option.
The exclusive reliance of steepest descents on gradients is both its weakness and its strength. Convergence is slow near the minimum because the gradient approaches zero, but the method is extremely robust, even for systems that are far from harmonic. It is the method most likely to generate a lower-energy structure regardless of what the function is or where it begins. Therefore, steepest descents is often used when the gradients are large and the configurations are far from the minimum. This is commonly the case for initial relaxation of poorly refined crystallographic data or for graphically built molecules. In fact, as explained in the following sections (on the conjugate gradients and Newton-Raphson methods), more advanced algorithms are often designed to begin with a steepest-descents direction as the first step.
It would be preferable to prevent the next direction vector from undoing earlier progress. This means using an algorithm that produces a complete basis set of mutually conjugate directions such that each successive step continually refines the direction toward the minimum. If these conjugate directions truly span the space of the energy surface, then minimization along each direction in turn must by definition end in arriving at a minimum. The conjugate gradients algorithm constructs and follows such a set of directions.
In conjugate gradients, hi+1, the new
direction vector leading from point i+1, is computed by adding
the gradient at point i+1, gi+1, to
the previous direction hi scaled by a
constant i :
This direction is then used in place of the gradient in Eq. 4-3, and a new line search is conducted. This construction has the remarkable property that the next gradient, gi+1, is orthogonal to all previous gradients, g0, g1, g2, ... , gi, and that the next direction, hi+1, is conjugate to all previous directions, h0, h1, h2, ..., hi. Thus, the term conjugate gradients is somewhat of a misnomer. The algorithm produces a set of mutually orthogonal gradients and a set of mutually conjugate directions.
Conjugate gradients is the method of choice for large systems because, in contrast to Newton-Raphson methods, where a second-derivative matrix (N (N + 1)/2) is required, only the previous 3N gradients and directions have to be stored. However, to ensure that the directions are mutually conjugate, more complete line search minimizations must be performed along each direction. Since these line searches consume several function evaluations per search, the time per iteration is longer for conjugate gradients than for steepest descents. This is more than compensated for by the more efficient convergence to the minimum achieved by conjugate gradients.

Figure 4-6. General Algorithm for Variants of
Newton-Raphson Method:
Another way of looking at Newton-Raphson is that, in addition to using the gradient to identify a search direction, the curvature of the function (the second derivative) is also used to predict where the function passes through a minimum along that direction. Since the complete second-derivative matrix defines the curvature in each gradient direction, the inverse of the second-derivative matrix can be multiplied by the gradient to obtain a vector that translates directly to the nearest minimum. This is expressed mathematically as:
The molecular energy surface is generally not harmonic, so that the minimum-energy structure cannot be determined with one Newton-Raphson step. Instead, the algorithm must be applied iteratively:
As elegant as this algorithm appears, its application to molecular modelling has several drawbacks. First, the terms in the Hessian matrix are difficult to derive and are computationally costly for molecular forcefields. Furthermore, when a structure is far from the minimum (where the energy surface is anharmonic), the minimization can become unstable. For example, when the forces are large but the curvature is small, such as on the steep repulsive wall of a van der Waals potential, the algorithm computes a large step (a large gradient divided by the small curvature) that may overshoot the minimum and lead to a point even further from the minimum than the starting point. Thus, the method can diverge rapidly if the initial forces are too high (or the surface too flat). Finally, calculating, inverting, and storing an N x N matrix for a large system can become unwieldy. Even taking into account that the Hessian is symmetric and that each of the tensor components is also symmetric, the storage requirements scale as (3N 2 ) for N atoms. Thus, for a 200-atom system, 120,000 words are required. The Hessian alone for a 1,000-atom system already approaches the limits of a Cray-XMP supercomputer, and a 10,000-atom system is currently intractable. Pure Newton-Raphson is reserved primarily for cases where rapid convergence to an extremely precise minimum is required, for example, from initial derivatives of 0.1 kcal mol-1 Å-1 to 10-8 kcal mol-1 Å-1. Such extreme convergence is necessary when performing vibrational normal mode analysis, where even small residual derivatives can lead to errors in the calculated vibrational frequencies.
Of the several different updating schemes for determining B, the two most common ones are the Broyden, Fletcher, Goldfarb, and Shanno (BFGS) and the Davidson, Fletcher, and Powell (DFP) algorithms.
Defining and
as the changes in the coordinates and
gradients for successive iterations, the approximate Hessians
(B-1 ) are given by the following in the DFP method:
The quasi-Newton-Raphson method has an advantage over the conjugate-gradients method in that it has been shown to be quadratically convergent for inexact line searches. Like the conjugate-gradients method, the method also avoids calculating the Hessian. However, it still requires the N x N disk space (N = number of atoms), and the updated Hessian approximation may become singular or indefinite even when the updating scheme guarantees hereditary positive-definiteness. Finally, the behavior may become inefficient in regions where the second derivatives change rapidly. Thus, this minimizer is used in practice as a bridge between the iterative Newton-Raphson and the conjugate-gradients methods.
To further increase the convergence rate of the conjugate-gradients minimization, the Newton equation that is solved for each line search direction is preconditioned with a matrix M. The matrix M may range in complexity from the identity matrix (M = I) to M = H. In the first limit, the Newton equation remains unchanged. However, in the second, there is no savings in computational effort, because M-1 H = I must be solved.
Figure 4-7. Flowchart of Truncated Newton-Raphson
Method:
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Minimization access
Minimization access
 Minimization - General Process
Minimization - General Process
 Minimization - General Methodology
Minimization - General Methodology
Copyright Biosym/MSI
{kind=link}
{kind=link}