Many of the data keywords are the same for all programs in this package. In all cases the meaning and token layout will always be the same for all programs which read the statement.
To simplify some of the descriptions below two abbreviations are used. The definition of the first is:
![]()
This construction is used to specify a particular residue of a particular chain. It is used both to refer to residues within a chain and to sequence entries within a chain type. The second form is used when one wishes to refer to a residue within the default chain (which has no name). For example, ``A|123'' designates residue 123 of chain A and ``123'' refers to residue 123 of the default (nameless) chain.
The second abbreviation is
![]()
This abbreviation is used when a particular atom is to be specified. If the atom belongs in the ``chain with no name'' the <Chain> field is left blank.
ASSUME RESIDUE_TYPE <Name> WHEN_CONTAINS N(<Name>)
This statement allows you to define a default residue type for any residue which contains all of the atoms listed, and no more. The puddle of water that is contained in most coordinate files presents a continuing maintenance problem in TNT's sequence file. Without the ASSUME statement you would have to edit the sequence file every time you added or removed solvent.
The following example will, I hope, clarify this statement's use. If your model has a great deal of DMSO, instead of creating individual RESIDUE statements for each one you could add to your control file the statement
ASSUME RESIDUE_TYPE DMSO WHEN_CONTAINS S C1 C2 O
Built into TNT is the assumption that a residue which contains only an atom named ``OH" is of type ``HOH". This is the PDB convention for water molecules.
ATOM <type> <X> <Y> <Z> <B> <Occ> <Atom designator> ATOMC <type> <X> <Y> <Z> <B> <Occ> <Atom designator> ATOMG <type> <X> <Y> <Z> <B> <Occ> <Atom designator>
These statements specify the parameters for the particular atom named
on the statement. The particular keyword used defines the coordinate
system in which
the position of the atom is specified. ATOM statements use fractional
coordinates, ATOMC statements use an orthogonal system (x is along ![]() --
this is not the same convention used in PDB format files.)
and ATOMG statements define the position in terms of map grid units. Internally,
positions are stored in the orthogonal system. To allow for the coordinate
conversion upon input, ATOM statements must be preceded by a CELL statement and ATOMG
statements must be preceded by both a CELL statement and a GRID statement.
For more information see Appendix
--
this is not the same convention used in PDB format files.)
and ATOMG statements define the position in terms of map grid units. Internally,
positions are stored in the orthogonal system. To allow for the coordinate
conversion upon input, ATOM statements must be preceded by a CELL statement and ATOMG
statements must be preceded by both a CELL statement and a GRID statement.
For more information see Appendix ![]() .
.
CELL <a> <b> <c> <Alpha> <Beta> <Gamma>
This statement gives the cell dimensions in Ångstroms and degrees. It is required in almost every program and giving one when not required does no harm so one should always give a CELL statement. Since ATOM and ATOMG statements require that a CELL statement proceeds them, it is best to always have the first statement in the data stream be a CELL statement.
CHAIN <Chain name> <Chain type> N(<Chain linkage>)
<Chain linkage> :==
<Residue name> <Residue designator> <Linkage type>
The CHAIN statement is used to specify the type of a particular chain and the locations and types of any links between this chain and other chains. The link is assumed to exist between the chain named on this statement and the nearest symmetry image of the target chain.
The link defined on this statement will most likely be disulfide bonds between separate polypeptides of an oligomer or connections to symmetry-related molecules.
For example, the statement
![]()
says that chain A is of type ALPHA and that there is a SS (disulfide) link between A|44 and B|123. The actual image of chain B linked to may be in the same asymmetric volume as A or it may be a symmetry image of B. The program will decide which symmetry operator to use.
For a more detailed description of this statement see the ``TNT Sequence File'' chapter in the TNT Users' Guide.
FORMFACTOR <Atom type> <Form factor parameters>
This statement allows the user specify a form factor approximation for a particular atom type. The form factors for all the atom types present in your structure must be defined each time you run a program which calculates electron density (or structure factors) from the model. A list of the definitions for many atom types is provided in the file $tntdata/formfactor.dat.
The atom type name on the statement can be up to 4 characters long and must match the atom type name on the ATOMx statements. The equation used to fit the form factors of the atoms is the sum of three gaussians,
![]()
where s is ![]() . On the FORMFACTOR statement the
numbers are given in the order of A, B, C, D, E, F. The first pair
should be the pair with the largest exponential coefficient (e.g.
B>D>F). If F is equal to zero then one has the form of the equation
used by Lee and Pakes (Acta Cryst. (1969). A25, 712) and the
parameters from their paper can be used. (The values used in TNT are not
from this paper, however there is no problem using the paper's values
together with the TNT numbers.) The temperature factor parameters, B, D,
and F, cannot be negative.
. On the FORMFACTOR statement the
numbers are given in the order of A, B, C, D, E, F. The first pair
should be the pair with the largest exponential coefficient (e.g.
B>D>F). If F is equal to zero then one has the form of the equation
used by Lee and Pakes (Acta Cryst. (1969). A25, 712) and the
parameters from their paper can be used. (The values used in TNT are not
from this paper, however there is no problem using the paper's values
together with the TNT numbers.) The temperature factor parameters, B, D,
and F, cannot be negative.
Here are some examples:
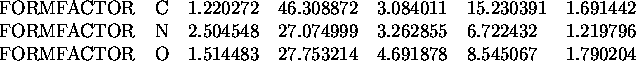
These statements define the form factors for atom types ``C'', ``N'', and ``O''. The parameter F is not given so it is assumed to be zero.
GEOMETRY <Residue type> <Restraint type> -
<Ideal value> <Sigma> N(<Atom name>)
GEOMETRY <Linkage type> <Restraint type> -
<Ideal value> <Sigma> N(<Atom name>)
GEOMETRY statements are used to define geometry restraints for particular residue or linkage types. (See the ``Standard Geometry Definition" chapter of the TNT Users' Guide.)
GRID <Grid X> <Grid Y> <Grid Z>
This statement gives the number of grid divisions per unit cell along the crystallographic axes x, y, and z. This line must be included if the coordinates are read in ATOMG format, or if an ATOMG coordinate file is to be created through the use of the PUNCH command. The GRID statement must precede any use of ATOMG statements.
LATTICE <Text>
This statement gives the name of the lattice centering operators. The codes
to be used are listed in Table ![]() .
.
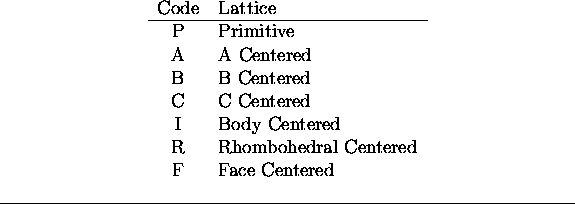
Table: Coded values for each lattice class
The default, if no statement is given, is Primitive.
The proper LATTICE and OPERATOR statements for all useful space groups are
given in the directory $tntsymmetry (TNT_SYMMETRY: on VMS).
The name of the file is simply
the name of the space group with a ``.dat'' appended. The file which
includes the definitions of the space group P ![]() is $tntsymmetry/p4322.dat.
is $tntsymmetry/p4322.dat.
While the LATTICE statement recognized R centered cells, TNT's FFT code will not perform Fourier transforms in this setting. Rhombohedral cells must be described in their hexagonal setting.
LINK <Residue name> <Target name> <Linkage type>
The link statement defines a stereochemical link between a residue in ``the chain with no name'' and a residue in a symmetry image of the chain. For instance, ``LINK 128 128 SS'' means that residue 128 is connected to a symmetry image of itself by a disulfide bond.
Additional information can be found in the ``TNT Sequence File" chapter of the TNT Users' Guide.
OPERATOR <Symmetry operator> [; <Operator name>]
The OPERATOR statement allows the input of the symmetry operators of the space group. One OPERATOR statement must be given for each symmetry operation, and the symmetry operator is given in the form used in the International Tables. Each operator may be given a name by the user. Because the crystal lattice is given on the LATTICE statement you should not enter the centering operators on OPERATOR statements. Only list the equivalent positions as listed in the International Tables.
To specify a name for an operator terminate the algebraic definition with a semicolon and enter the name at the end of the line. If the user does not do this the program will select a name for the operator. Here are some examples:
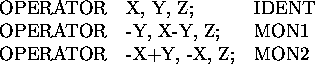
The proper LATTICE and OPERATOR statements for all useful space groups are
given in the directory $tntsymmetry (or
TNT_SYMMETRY: on VMS).
The name of the file is simply
the name of the space group with a ``.dat'' appended. The file which
includes the definitions of the space group P ![]() is
$tntsymmetry/p4322.dat.
is
$tntsymmetry/p4322.dat.
OPTION {SET | CLEAR} N(<Option name>)
This statement either sets or clears the options named. Different programs have different options which can be turned on or off by this mechanism. By default all options are cleared when the program starts.
The only option recognized by all programs is VERBOSE. When this option is SET the program will write detailed information to the standard output.
RESIDUE <Residue designator> <Residue type> -
N(<Residue linkage>)
<Residue linkage> :== <Residue name> <Linkage type>
This statement defines the residue type of a particular residue within a chain type along with the targets and types of any linkages to other residues within the same chain. For a more detailed description of this statement the ``TNT Sequence File'' chapter of the TNT Users' Guide.
RESOLUTION <Inner limit> <Outer limit> RESOLUTION <Outer limit>
This statement specifies the inner and outer resolution limits of the crystallographic data in Ångstroms. The order of the resolution limits on the statement is unimportant. If the inner limit is not given it is presumed to be equal to infinity, which means that all data from F(000) to the outer limit will be used.
TRANSFORMATION <Name> -
{[{EULERANGLES 3<Values> TYPE {CROWTHER | HUBER | ROSSMANN} |
POLARANGLES 3<Values> TYPE {KABSCH | ROSSMANN} |
MATRIX 9<Values> |
AXIS <Angle> 3<Direction Cosines> }]
[CENTER 3<Values>]
[SHIFT {XYZ 3<Values> | B <Value> | OCC <Value>}]
SYSTEM {TNT | PDB | FRACTIONAL | GRID}
|
OPERATOR <Equivalent Position>}
Each transformation contains a rotation, a center, a translation, and a coordinate system. When the transformation is defined with the OPERATOR modifier all of them are defined at once. With the other modifiers the parts are defined separately. Each part which is undefined is assumed to be 1) a zero degree rotation, 2) centered at the origin, 3) no translation. In an attempt to avoid confusion the angular TYPE and coordinate SYSTEM cannot be defaulted.
In all cases the order of application is 1) the rotation about the center and 2) the translation.
A transformation with a particular name can only be defined once. If the determinate of the MATRIX is not equal to one a warning will be generated but the transformation will be accepted even though this will usually be mistake.
The Crowther Euler angles are defined as a rotation about the Z axis,
followed by a rotation about the new Y axis, and finally a rotation
about the ultimate Z axis. The form of the rotation matrix is
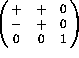 .
.
The Huber Euler angles are defined as a rotation about the Z axis,
followed by a rotation about the new Y axis, and finally a rotation
about the ultimate X axis. The form of the rotation matrix is
.
The Rossmann Euler angles are defined as Z, X, Z, with the form
.
In Kabsch polar angles the first two angles define the direction of an axis and the third angle defines a rotation about that axis. The first angle is the angle between the X axis and the projection of the rotation axis onto the XY plane. The second angle is the angle between the Z axis and the rotation axis. The first angle may have any value. The second angle must lie between 0 and 180 degrees. The third angle may also have any value.
In Rossmann polar angles the first two angles define the direction of an axis and the third angle defines a rotation about that axis. The first angle is the angle between the X axis and the projection of the rotation axis onto the XZ plane. The second angle is the angle between the Y axis and the rotation axis. The first angle may have any value. The second angle must lie between 0 and 180 degrees. The third angle may also have any value.
WEIGHT N(<Restraint class> <Value>)
WEIGHT statements are used to specify a weight for each module. Some modules, like that for stereochemistry, will accept a weight for each of several different classes of restraints, such as bond lengths and bond angles.