Issue the command biosym_tutorial -i at the system prompt. If the Discover tutorial files are not installed, enter discover at the first prompt.
Additional information on installing tutorials.
Press <Enter> to get out of the installation menu, then enter 3 to change to the tutorial directory, followed by entering 1 to start up the Insight II program.
It takes a few moments for the Insight program to start up.
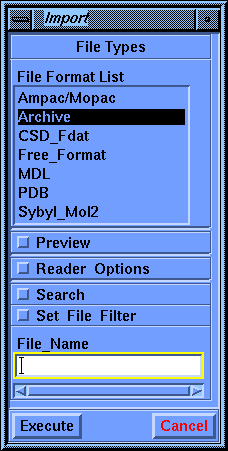
Select the File/Import command and fill in the File_Name parameter box with pp.car, by choosing it from the value-aid. Scroll the list, if necessary, by using the scroller on its right side. Select Execute.
The molecule that appears corresponds to a fragment of isotactic polypropylene, with the same stereochemistry for the methyl side chains. If you wish, after completing this tutorial you could modify or rebuild the fragment to correspond to syndiotactic polypropylene, having the methyl groups on opposite sides of the main chain. You may select Cancel to put away the parameter block so you can see the molecule better. You can use the mouse or dials to move the molecule, if you want.
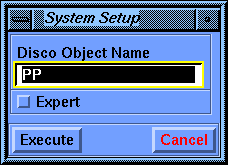
Select the Molecule/Label command. Pick the molecule to set Molecule Spec to PP, if necessary, and Execute the command.
Each atom of the molecule in the display area becomes labelled. This will aid in later definition of the torsion angles.
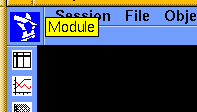
Go to the Module pulldown (that is, click the Biosym logo) and select Discover_3 from the list that appears.
The Discover_3 pulldowns appear on the lower menu bar.
This defines PP as the system that you want to work with.
The Discover_3 commands function a bit differently than most of the commands in the Insight interface. Since you are setting up a perhaps lengthy and complex calculation, the commands store the information you enter. The calculation does not start until you select the D_Run/Run command. Thus, nothing actually happens when you execute all of the other commands in the Discover_3 module except that the information is stored. If you make a mistake or forget something, you can come back and add, delete, or modify anything in the setup for the job at any time until you finally run the job with the D_Run/Run command. The last part of this tutorial shows you how to add a step that you forgot.
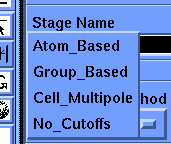
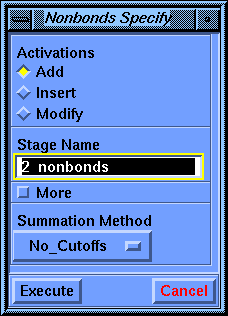
Select the Specify/Nonbonds command. When the parameter block appears, set Summation Method to No_Cutoffs (click the box under Summation Method and choose No_Cutoffs from the list that appears) and select Execute.
The Discover program uses the cell multipole method by default for nonbond calculations. This is the method of choice for most molecules, but since this tutorial is working with such a small molecule you can ask the Discover program to simply calculate all the pairwise interactions. This is the fastest and most accurate method for molecules with fewer than about 500-1000 atoms, depending somewhat on what type of computer you are using.
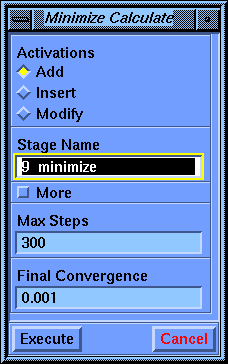
Choose the Calculate/Minimize command. The default parameter values are sufficient, so select Execute.
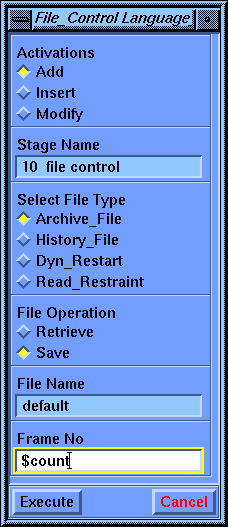
Select the Language_Control/File_Control command. Set the File Operation parameter to Save. For Frame Number, enter $count. Select Execute to store this stage.
Notice that the File_Control command requires a Frame Number to control writing to the archive file. If you entered a constant, such as 1, all the results would be written to the same frame, overwriting all previous results. That is why you were instructed to enter a variable name ($count). However, that variable has not been initialized or incremented for each new structure. Therefore you need to insert a couple of earlier stages into the calculation to take care of this.
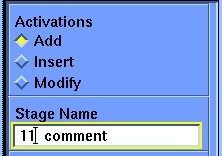
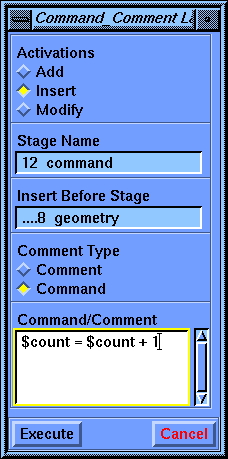
Select the Language_Control/Command_Comment command.
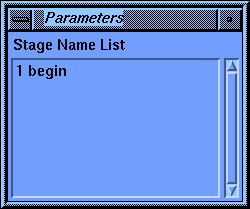
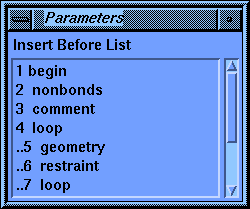
The first stage control parameter, Activations, has three choices: Add, Insert, and Modify. Beneath the second parameter, Stage Name, is the default name for this stage. When the parameter block first comes up the default choice for Activations is Add, and the Stage Name parameter is active. You may change the Stage Name parameter if you wish, but the default is usually the best choice. The default shown now is 11 comment. The number is the position of this stage in the list--eleventh in this case. The rest of the name (comment) tells you what this stage does. You may have noticed that you haven't created a comment stage before now, yet another one (1 begin) claims to be the first one (according to the list in the value-aid). The reason for this is that the Discover program automatically starts every calculation with a comment, so the very first stage in any calculation is a comment. If you use the default scheme for stage names, the Discover program maintains them for you.
Try setting the Activation parameter to Modify. The Parameters value-aid that appears lists all the stages of the same type (comments in this case). If you needed to revise some of the parameters in one of the stages, you would select the stage's name in the value-aid. The parameter block would reflect the settings of all the parameters in that stage. After changing anything that you needed to, you would select Execute to save the modified stage.
You need to insert a stage, however, so:
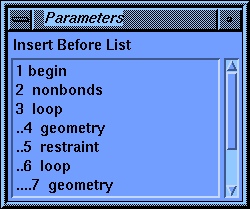
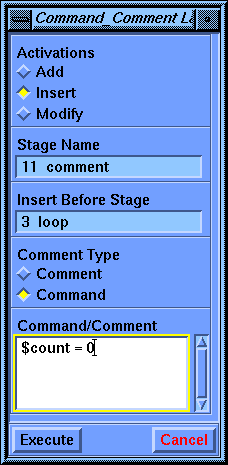
Set the Activations parameter to Insert. The stage name should be 11 comment. Now click the Insert Before Stage parameter box to activate it.
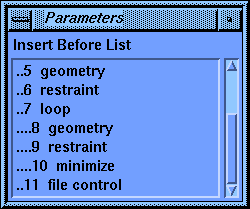
The value-aid lists all the stages that you have created so far. Notice that each nested level of the loops is indented by a pair of periods to help you see the structure of your calculation.
You need to initialize the counter before the first loop:
Choose the 3 loop stage in the value-aid. Set the Comment Type parameter to Command and then type $count = 0 in the Command/Comment parameter box. Note the spaces on either side of the equals sign. Select Execute to create this stage.
Notice in the Parameters value-aid that the third stage is 3 comment and the numbers for subsequent stages have been incremented by one.
Still in the Command_Comment parameter block, check that the Activations parameter is still set to Insert. Select the Insert Before Stage parameter if it is not already active. From the value-aid, select the entry ....8 geometry. Then change the Command/Comment parameter to $count = $count + 1. Select Execute to save this stage.
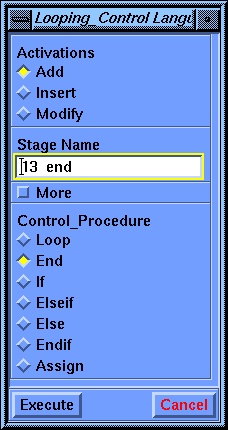
Select the Language_Control/Looping_Control command. Set the Control Procedure to End. Select Execute to end the inner loop over the psi torsion angle.
Select Execute a second time to end the outer loop over the phi torsion angle.
You have completed the setup for the job, but before you run the calculation it would be a good idea to look over the setup to make sure that you haven't forgotten anything. There are several ways to do this. You could look back through all the stages using the Modify Activations parameter to look at the settings in the menus for each stage. But it is probably easier to look at the input file for the program, so let's do that:
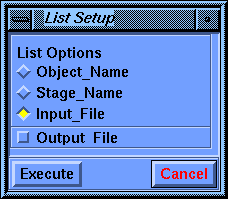
Select the Setup/List command, select Input_File as the List Options parameter, and Execute the command.
The textport pops in front of the Insight window and should contain a listing similar to the annotated input file.
The atom names in the molGeom and restraint commands might be different, if you built the molecule instead of using the one that was provided or if you defined the torsions differently than in the instructions. Other than that, the listing of your input file should be identical to the example file.
Check the listed file carefully, especially noting the use of the $phi and $psi variables. You should notice that each command in the input file corresponds to one of the stages that you set up with the parameter blocks, except that a writeFile coordinate filename = .cor line was added by the Discover program. The writeFile command is always added after minimizations to write out the .cor file.
After comparing your input file with the example file, select the Textport button to put the textport away.
If your input file does not correspond to the annotated example file (except, of course, for the annotations themselves), you need to correct the errors. If some of the parameters for a command are not correct, you need to modify that stage, as was outlined above. If one of the commands is missing completely, you accidentally skipped over it when creating the stages. In this case, you need to go back and insert the appropriate stage in the correct place. Follow the same procedure that you used above to insert the Command/Comment stage, but of course insert a stage of the correct type.
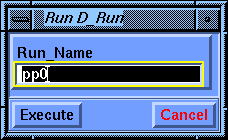
Select the D_Run/Run command and be sure that pp0 is the value of the Run_Name parameter. Execute the command to start the calculation.
The calculation will take anything from about 10 minutes on a fast workstation (such as an R4000-based SGI or IBM RS-6000 370) to a few hours on slower machines (such as an SGI 4D-25). If you leave the Insight interface running while the Discover job executes, you can watch the progress of the run in the display area, and you will be notified when the job is completed. (If you want to check whether the job has finished, you can also select the Background_Job/Completion_Status command and select Execute. A message on the job's status appears in the information area near the bottom of the Insight screen.) If you quit the Insight interface, you can use commands such as gr_top on the SGI, smit on the IBM, or ps on either machine to check if the job is still running. After it completes, you are ready to read the results back into the Insight environment and generate a phi-psi map.
If you exited the Insight environment, restart it as above and use the File/Import command to read in the 2,4-dimethylpentane molecule you used previously. This molecule was saved in a file called pp0.car when you started the Discover calculation.
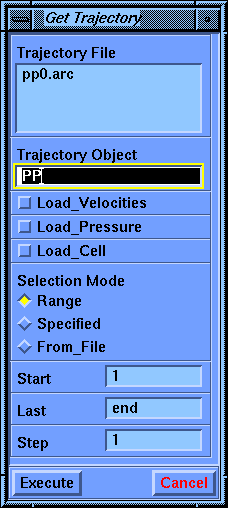
Go to the Analysis module by clicking the Biosym logo. Select the Trajectory/Get command from the lower menu bar, choose pp0.arc in the value-aid, set the Trajectory Object to PP, and select Execute to read in the archive file.
In the information area at the bottom of the Insight window, you should see a message that trajectory records for 23 atoms and 169 images were processed. If fewer frames were found, your job was either not completed or terminated early for some reason. You can look in the pp0.out file for error messages.
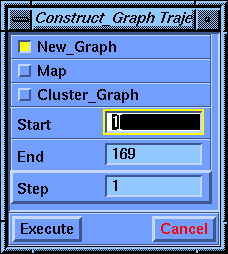
Select the Trajectory/Construct_Graph command. Select Execute.
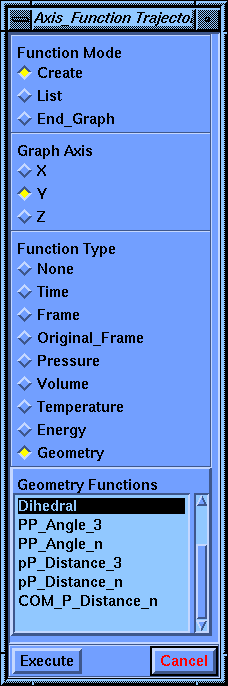
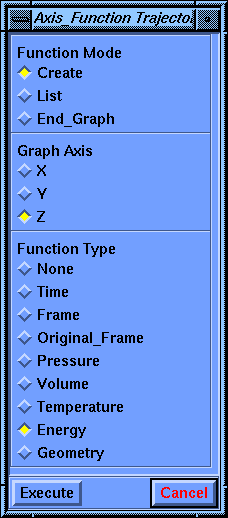
Another parameter block, called Axis_Function Trajectory, appears for defining the graph.
Choose Geometry as the Function Type and select Dihedral in the scrolling list that appears. Select Execute.
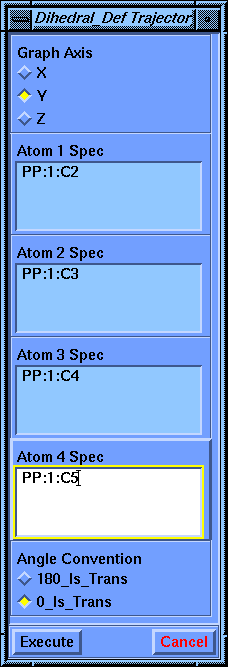
A Dihedral_Def Trajectory parameter block appears for defining the dihedral.
Choose 0_Is_Trans for the Angle Convention, then pick the first four carbon atoms in the backbone (C1 through C4, in that order). (You can type in these values or move the molecule if they are difficult to pick.) This fills in the Atom 1 Spec through the Atom 4 Spec parameters. The command automatically executes when you pick the fourth atom.
When the Axis_Function Trajectory parameter block reappears, the default Graph Axis has changed to Y and Dihedral is still selected. Select Execute.
When the Dihedral_Def Trajectory parameter block appears, choose 0_Is_Trans for Angle Convention. Now select the second through fifth carbon atoms in the backbone. Again the command automatically executes when you pick the last atom to fill in the Atom 4 Spec parameter.
The Axis_Function Trajectory parameter block appears again, this time with the Z axis chosen. Choose Energy for the Function Type and then select Execute.
An Energy_Def Trajectory parameter block appears for defining what type of energy to plot. The default is Total for Energy Type. Check that it is indeed chosen, and then select Execute.
When the Axis_Function Trajectory parameter block reappears, choose End_Graph for the Function Mode and select Execute.
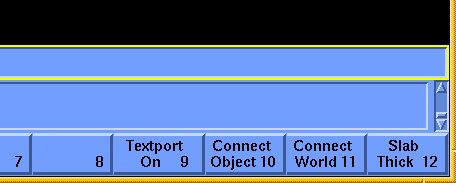
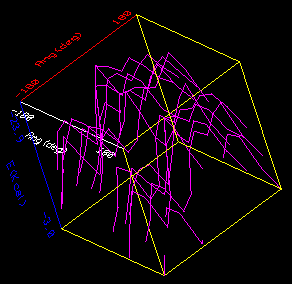
A 3D plot appears in the Insight window. Initially you are looking down the Z axis, so it appears 2D. If you want to rotate and move the graph, press the <F10> key (or equivalently select the on-screen button labeled Connect Object with the left mouse button). Now pick one of the corners of the graph with the left mouse button. You should see a message in the information window at the lower part of the Insight screen saying that the dials are connected to GRAPH1. You may need to be quite accurate in picking the corner of the graph, so try again if you do not see the message in the information window. After you are connected to the graph, you can rotate it by dragging with the right button held down. You translate it by dragging with the center button down and resize by dragging with both the center and right buttons down. To resize, dragging up on the screen enlarges the graph, down shrinks it. If part of the graph appears to be cut off, you may need to increase the slab thickness with the Transform/Clip command and/or translate the graph along the Z axis with the dialbox.
You will use a spectrum to color the phi-psi map.
You can shrink and position the spectrum in a convenient place in the same way that you manipulated the graph in the previous step.
Select the Graph/Contour command from the lower menu bar. Choose Range_Create from the popup under Contour_Action_2D. Toggle the New_Graph parameter to off. The Graph Name should be GRAPH1, and Plot Spec should be GRAPH1:1. If they are not, correct them. Toggle Use_Spectrum to on and set the Spectrum Name to ENERGY, the name you gave the spectrum that you created.
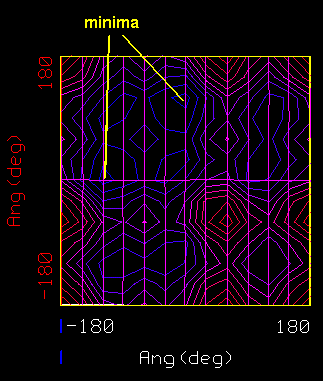
MAP1:p3.0:60 Contour level: 3.00, X=99.28, Y=213.04, Z=0.00
appears in the information window. The Contour Level is the energy
of the contour in kcal mol-1. The minimum for this
system is at about -23.9 kcal mol-1 at a
gauche-trans conformation.
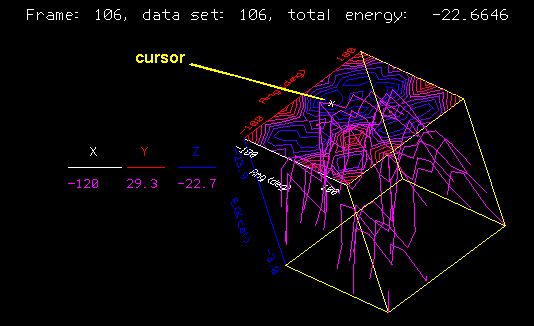
Select the Trajectory/Animate command and select Execute.
The displayed molecule starts to cycle through the archived structures. You can cancel the parameter block to see more of the display area. The molecule's structure is updated, information on the trajectory frame and energy is displayed, and a cursor moves along the graph to indicate the position of that structure in the graph. The <F8> key (or the button now labeled Anim Frame) toggles between automatic playback and a manual mode. In the manual mode, you click the dialbox labeled Anim Frame in the lower left portion of the Insight window. Selecting the left side of the button moves a frame backward; the right side, forward. The frames cycle in the direction you select for as long as you hold the mouse button down.
You should see a pair of minima in the contour map on either side of the diagonal in the upper left quadrant. One is at -120,0; the other 0,120. These correspond to the two symmetric conformations gauche-trans and trans-gauche. Because we truncated the polypropylene to a very symmetric dimethylpentane, these two structures are in fact identical: they interchange the methyl groups that terminate the backbone with the methyl groups that are the sidechains. If we had used a longer segment of the polymer--five or six monomers, say--rotating and restraining all the phi angles as a group and all the psi angles also as a group, the two minima would have become slightly different. One corresponds to a left-handed helix repeating after three monomers; the other, a right-handed helix. Because of the chirality due to the side chains, which are all R or all S in isotactic polypropylene, one of the minima has a lower energy than the other.
Type quit on in the command line near the bottom of the Insight screen and press <Enter>.
Now enter 5 to exit the biosym_tutorial script.
 Main
access page
Main
access page  Insight UIF access
Insight UIF access
 Insight UIF - Tutorial access.
Insight UIF - Tutorial access.
 Phi-Psi Map Lesson, Introduction
Phi-Psi Map Lesson, Introduction
 Pre-recorded Script Lesson, Introduction
Pre-recorded Script Lesson, Introduction
Copyright Biosym/MSI
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}