- Invoking the Insight II program
Issue the command biosym_tutorial -i at the system prompt. If
the Discover tutorial files are not installed, enter
discover at the first prompt.
Additional information on installing
tutorials.
Press <Enter> to get out of the installation menu, then enter
3 to change to the tutorial directory, followed by entering
1 to start up the Insight II program.
It takes a few moments for the Insight program to start up.
- Setting the display defaults for
molecules
To make molecules easier to work with, you can set the default
display features to show double and partial double bonds and to color
atoms according to their elements:
- Select the Molecule/Set_Defaults
command (from the upper menu
bar) and set the
following parameters:
-
File Types - All
Coloring Method - By_Atom
Default Bond Order - on
Default DepthCueing - on
Default Line Width - 1 or 2
Select Execute.
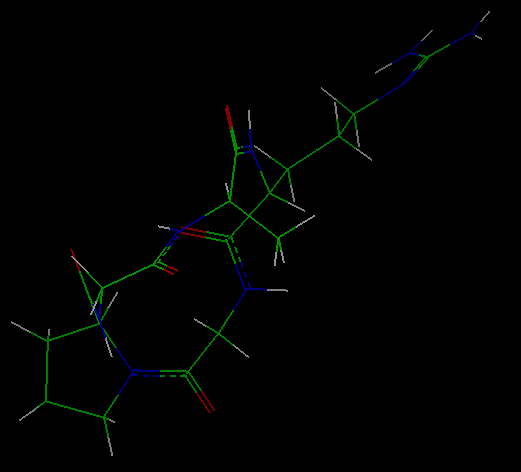
- Reading in the cyclic peptide molecule
Select the File/Import
command. Fill in the File_Name parameter box with cyclo0.car by
choosing this filename from the Files value-aid (you may have
to scroll the list by using the scroll bar on the side of the
value-aid). Execute the
command.
The molecule that appears
was constructed by forming a (distorted) bond between the first and
last residues of the linear peptide gly-pro-gly-ala-arg.
You can use the mouse buttons to translate and rotate the molecule to
facilitate viewing it.
- Activating the Discover 95.0 program
The peptide can be minimized to remove the strain caused by the
long bond. The following calculation is a simple minimization that
uses the default parameter values.
Go to the Module pulldown (that is, click the Biosym logo) and select
Discover_3 from the list that appears.
The Discover_3 pulldowns appear on the
lower menu bar.
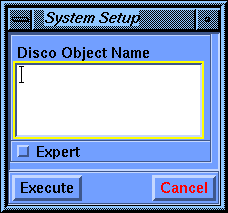
- Specifying the system to be used in the
calculation
Select the Setup/System
command.
Pick the peptide structure in the display area or choose CYCLO0 from
the Assem/Mol Names value-aid to fill in the Disco Object
Name parameter box.
Select Execute.
This defines CYCLO0 as the system that you want to work with and
also enables you to execute additional Discover_3
commands.
- Setting up the minimization
- Choose the Calculate/Minimize
command and check that the following default values are set:
- Activations - Add
Stage Name - 2 minimize
More - off
Max Steps - 300
Final Convergence - 0.001
Select Execute.
- Specifying the output
Choose the Analyze/Output command. Specify the Stage
Name by typing 2 minimize in the box or choosing 2
minimize from the Stage Name List value-aid near the
parameter block.
- Select the following
options:
-
Delete_File_Type - off
File Type - Output
Discover File Name - default
Before - on
During - off
After - on
Instant - off
Running - off
Batch - off
Energy_Output - on
Total_E - on
Kinetic_E - off
Potential_E - off
Total_Nonbond_E - on
Coulomb_E - on
VdW_Total_E - on
VdW_Repulsion_E - on
VdW_Dispersion_E - on
Hydrogen_Bond_E - off
Total_Internal_E - on
Bond_E - on
Angle_E - on
Torsion_E - on
Out_Of_Plane_E - on
Total_Cross_Term_E - off
Temperature_Output - off
Select Execute.
- Running the minimization
Select the D_Run/Run command
and be sure that cyclo00 is the value of the Run_Name
parameter. Execute the command to start the calculation.
At any time after this step, you may compare your input file with an annotated copy of the input file, by going
to a different shell window (i.e., not the one from which you started
the Insight program) and using the UNIX cd command to go to your
tutorial directory, then the more command to list the cyclo00.inp
file.
The conformation of the molecule is updated as the run proceeds,
and information on the progress of the run is printed in the display
area.
When the job is finished (less than a minute on a 100-MHz SGI Indigo2
IRIX 5.2) a window appears, indicating
that the job has completed. Click the Continue button to put
the window away.
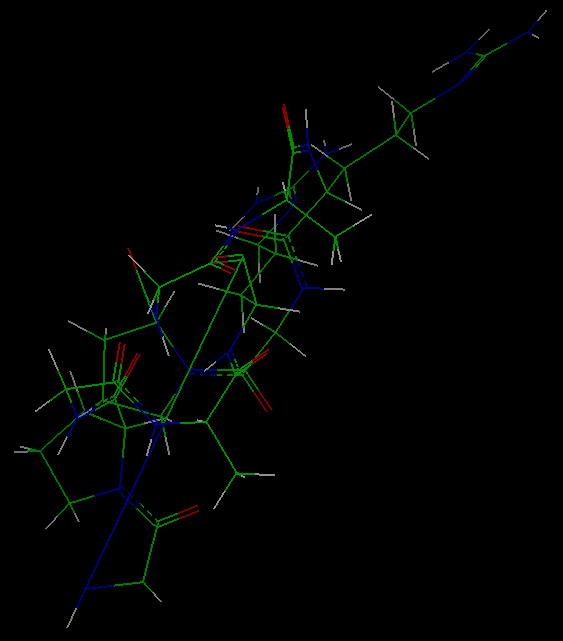
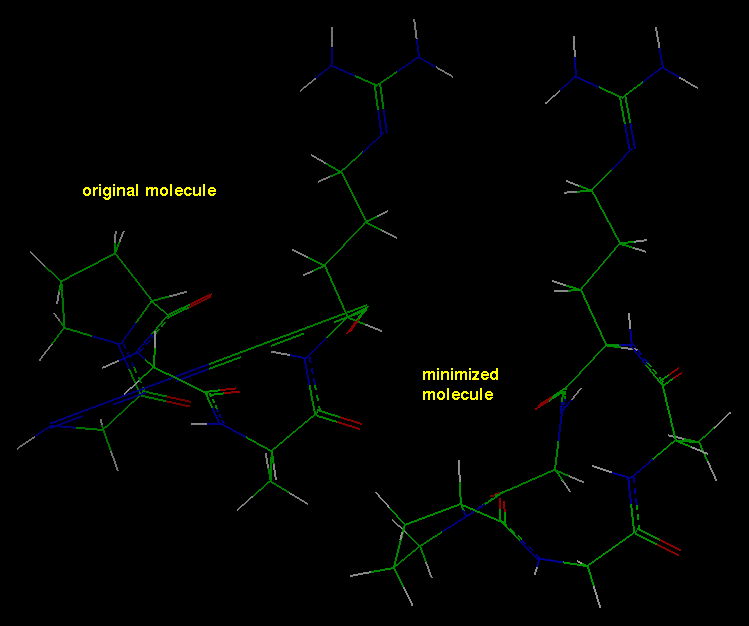
- Comparing the optimized and initial
structures
After the calculation has finished, the optimized structure (now shown in
the display area) can be compared with the initial structure:
Select the File/Import
command from the upper menu bar. Specify cyclo0.car as the value of
the File_Name parameter and Execute the command.
This brings the original structure into the Insight display area
(probably on top of the minimized structure).
Connect the mouse to either of the molecules (use the Connect Object
button) to move it without moving the other. Move the molecules around
to compare their
configurations--note that the long bond in the original molecule
is now indistinguishable in length from the other bonds in the
optimized molecule.
- Determining the energy of the optimized
structure
To check the values of the optimized energy and its components, select
the Session/Unix command
and select Execute. Wait until a shell window and a UNIX prompt
appear, then list the output file by entering the following command at
the UNIX prompt (represented by >):
> more cyclo00.out
You may compare your output file with an annotated copy of the output file.
Note that the default calculation uses group-based nonbond energy cutoffs
of 9.50 Å for both the van der Waals and Coulombic
interactions.
After viewing the output file, return to the Insight
environment by typing exit at the UNIX prompt and then
selecting the Textport
button.
This completes the illustration of a simple minimization.
- Removing the original structure from the
display area
In the next part of this lesson, we need only the minimized
structure. Since the initial (non-minimized) structure is no longer
needed, it should be removed:
Select the Object/Delete
command from the upper menu bar. Specify the original structure (now
called CYCLO01) and select Execute.
- Proceeding to the next lesson
Do not quit the Insight program, but proceed to the lesson on Simple Dynamics with
Equilibration and Data Collection Stages or to its introduction.
 Main
access page
Main
access page  Insight UIF access
Insight UIF access
 Insight UIF - Tutorial access.
Insight UIF - Tutorial access.  Minimization Lesson, Introduction
Minimization Lesson, Introduction
 Simple Dynamics Lesson, Introduction
Simple Dynamics Lesson, Introduction 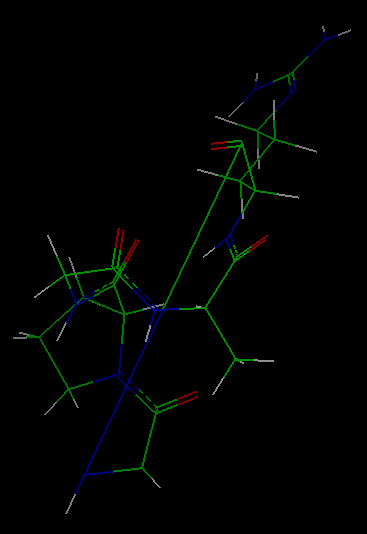{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}