 Minimization - General
Methodology Minimization - General
Methodology
Minimization - General
Methodology Minimization - General
MethodologyFor simulation strategies that involve minimization, several considerations must be addressed, including:
The minimized energies calculated for enzyme-substrate complexes can be used to estimate relative binding enthalpies, but there are two caveats. First, for a meaningful comparison of the relative binding of two different substrates, a complete thermodynamic cycle must be considered (Kirkwood 1935, Quirke and Jacucci 1982, Tembe and McCammon 1984, Mezei and Beveridge 1986).
In practical terms, this means that an enthalpy calculation for the various substrates in water must be made. Where the relative binding of two different enzymes to the same substrate is calculated, the energy of each enzyme with solvent in the binding site must be calculated.
A second consideration for using minimization results to estimate relative binding strengths is that the entropy is neglected in such calculations. Direct calculation of entropy differences is a computationally intensive process, and only recently has it been taken into account correctly by calculations of relative free energies (Hagler et al. 1979c, Hwang and Warshel 1987, Warshel et al. 1986, Singh et al. 1987, Straatsma et al. 1986).
The extent of the errors introduced by neglecting entropic contributions in the simpler minimization calculations is difficult to estimate, although, as with the zero-point energy, the entropy can be estimated for a system small enough that its normal mode frequencies can be calculated (Hagler et al. 1979c).
The relative importance of these fundamental considerations depends on the objective of the calculation. When studying the relative binding in an enzyme active site of two substrates, one of which is flexible and the other rigid, entropic effects may be crucial for obtaining even qualitative agreement with experimental binding constants. On the other hand, if a putative compound overlaps sterically with many active-site atoms and causes hundreds of kilocalories of strain energy even in a minimized structure, the compound can be rejected confidently. The bottom line is that physical-chemistry common sense cannot be abandoned when you are setting up a calculation and interpreting the results.
Several practical aspects of the conjugate gradients method are worth mentioning. First, the conjugate gradients algorithm requires convergence along each line search before continuing in the next direction. The gradient at step i+1 must be perpendicular to hi or the derivation guaranteeing a conjugate set of directions breaks down. Second, to start conjugate gradients, an initial direction h0 must be chosen that is equal to the initial gradient. Finally, additional storage is required for an extra vector of N elements to hold the N components of the old gradient. For energy minimization in Cartesian space this would be the 3N derivatives of the energy with respect to the x, y, and z coordinates of each atom. This makes conjugate gradients the method of choice for systems that are too large for storing and manipulating a second-derivative matrix, as is required by Newton-Raphson style minimizers.
Also, note that the derivation invokes a quadratic approximation. For nonharmonic systems, conjugate gradients can exhaustively minimize along the conjugate directions without converging. This condition generates an error message from the Discover program that the minimizer may have gotten stuck at a saddle point. If this occurs, you can restart the algorithm. Several minimizations may be required. For a detailed discussion of this algorithm, see the excellent text by Press et al. (1986) or the somewhat more formal treatment by Fletcher (1980).
Another example of the use of restraints is in modeling incomplete systems. Often, it is difficult or impossible to construct a realistic environment around parts of a model system. Only a partial structure of a large protein complex may be available, and some atoms must be restrained to stay near their initial crystal positions because they do not ``feel'' interactions with neighboring (missing) amino acids, membrane, or solvent. If the site of interest (for instance, a binding site for a competitive inhibitor) is well characterized but other parts of the enzyme are unknown or would require too much computation time if they were included, a limited study can still be carried out with the ends tethered to their crystal coordinates. Usually, these restraints are permanent parts of the model. The results of such calculations must be critically evaluated but can be valid if the ligand binding does not depend on interactions with missing pieces of the model or on conformational flexibility in the tethered regions.
As a final example, tethering can be used to gently relax a crystal structure. Often, crystal coordinates, even if highly refined, have several strained interactions due to intrinsically disordered or poorly defined atomic positions, which, upon minimization, give rise to large initial forces. If these forces are not restrained, they can result in artifactual movement away from the original structure. The general approach is to progressively relax parts of the model in stages, starting with the least well determined atoms, until the entire system can minimize freely. The restraints are ultimately removed so that the final minimum represents an unperturbed conformation. It is usually not necessary to minimize to convergence at each stage-the object is to relax the most-strained parts of the system as quickly as possible without introducing artifacts.
A typical approach to relaxing a crystal-derived protein system would be:
In a molecular minimization, the atomic derivatives may be summarized as either an average, a root-mean-square (rms) value, or the largest value. The average, of course, must be an average of the absolute values of the derivatives, because the distribution of derivatives is symmetric about zero. A rms derivative is a better measure than the average, because it weights larger derivatives more, and it is therefore less likely that a few large derivatives would escape detection, which can occur with simple averages. Nevertheless, regardless of whether you choose to report convergence in terms of the average or rms values of the derivatives, you should always check that the maximum derivative is not unreasonable. There can be no ambiguity about the quality of the minimum if all derivatives are less than a given value.
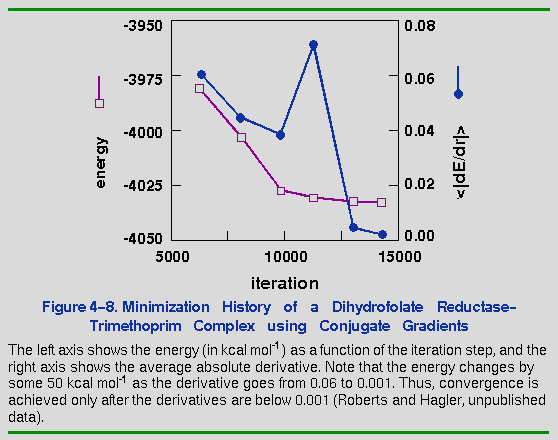
The more difficult question is, What value of the average or rms derivative constitutes convergence? The specific value depends on the objective of the minimization. If you simply want to relax overlapping atoms before beginning a molecular dynamics run, minimizing to a maximum derivative of 1.0 kcal mol-1 Å-1 is usually sufficient. However, to perform a normal mode analysis, the maximum derivative must be less than 10-5, or the frequencies may be shifted by several wavenumbers. Figure 4-8 shows the minimization history of the DHFR-trimethoprim protein-substrate system, which required some 14,000 iterations to converge to an average derivative of 0.0002 kcal mol-1 Å-1 (Roberts and Hagler, unpublished data). Often, minimizations of protein systems are stopped when average derivatives between 0.02 kcal mol-1 Å-1 and 0.5 kcal mol-1 Å-1 are achieved. If this simulation had been terminated when the average derivative had reached just 0.02, the structure would have been 30 kcal mol-1 higher in energy and approximately 0.3 Å (rms) away in structure from the final minimum (Roberts and Hagler, unpublished data). This difference represents 25% of the total movement during minimization. Thus, if quantitative measurements and comparisons of macromolecular structures are to be reliable, it would appear from this result that it may be necessary to allow minimizations to converge to average derivatives on the order of 0.002.
It is instructive to note that, although the energy must decrease monotonically during a minimization, the derivative need not. Figure 4-8 shows how the slope of a function can increase in magnitude during a traversal of the surface. This often occurs for functions as complex as molecular energy surfaces.
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Minimization access
Minimization access
 Minimization Algorithms
Minimization Algorithms
 Example Minimization Calculation
Example Minimization Calculation
Copyright Biosym/MSI
{kind=link}