 Minimization - Example
Calculation Minimization - Example
Calculation
Minimization - Example
Calculation Minimization - Example
Calculation
Binding of ligands to enzymes depends strongly on solvent and ionic interactions. Therefore, one of the first steps is choosing the ionization state of side chains and the treatment of solvent. In this study, the side chains of Glu, Asp, Arg, and Lys were charged, as were the N- and C-terminal amine and carboxylate groups. In addition, a His at position 45 is known to be protonated when NADPH is bound, so this amino acid was treated as a special case. Waters that appeared to play a structural role in the protein structure and any other crystallographic waters within 7 Å of trimethoprim and NADPH were retained. Additional waters were added to fill the volume within 7 Å of trimethoprim and NADPH and within 3 Å of all charged residues. All hydrogens were included explicitly, and a dielectric constant of 1 was used throughout.
A common and important modeling objective is to relax poorly defined regions of a protein structure without disrupting well defined regions. Above, we explained how template forcing and tethering can be used for this. In this study, the protein was tethered in stages to relax the least well refined parts of the model first, starting with the solvent. The backbone of the protein was restrained next to allow the side chains to make minor adjustments. Eventually, the entire system was relaxed unrestrained, until the minimization converged (at an average absolute derivative of 0.0002 kcal mol-1 Å-1).
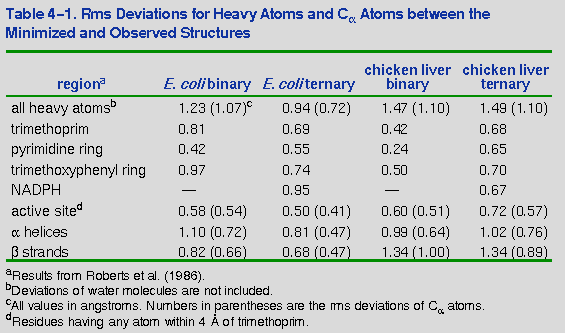
Table 4-1 summarizes the results
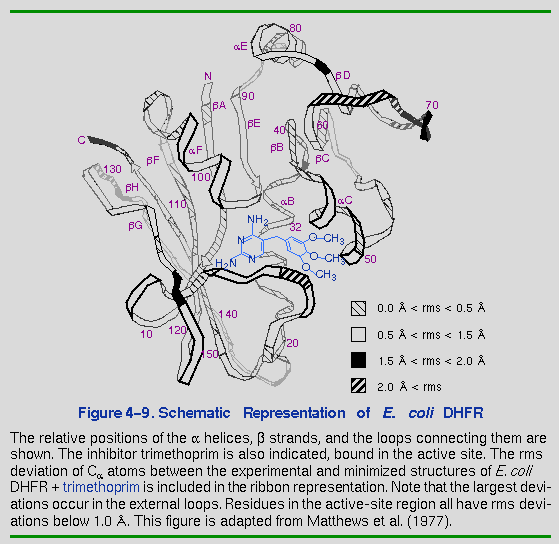
of several rms deviation analyses for each of the DHFR complexes. Figure 4-9 graphically
represents the rms deviations for the E. coli DHFR +
trimethoprim system. As expected, internal regions of the protein,
such as the active site, were found to fit the experimental structure
more accurately than areas on the surface of the protein. The well
defined areas of the protein, such as the active site and the C carbons of the
helices and
strands, show rms deviations substantially lower than does the overall
protein. The pyrimidine ring of trimethoprim, deep in the active site
cleft, deviates from the experimentally observed structure less than
does the trimethoxyphenyl moiety, which extends out towards the
surface of the protein. Similarly, in the chicken liver ternary
complex, the nicotinamide ring, which extends into the active site,
has a much smaller rms deviation (0.34 Å) than the NADPH
molecule as a whole, which generally lies on the surface of the
protein. Thus, the crucial area for this study, the active site, shows
good agreement with the experimental structure and was judged adequate
for further detailed structural and energetic analysis.
The selectivity of bacterial versus vertebrate DHFR for trimethoprim binding depends partially on cooperativity with respect to NADPH. Specifically, Baccanari et al. (1982) showed that the E. coli DHFR-trimethoprim dissociation constant is 40-fold less in the presence of NADPH, whereas for rodent lymphoma DHFR the corresponding decrease is only a factor of 2.8. By examining the two DHFR systems both with and without NADPH, the energetic basis underlying any cooperativity was investigated. Initially, two general mechanisms for cooperativity appeared possible: an indirect mechanism, in which the NADPH changes the protein so that it has higher affinity for trimethoprim; or a direct mechanism, in which favorable interactions between NADPH and trimethoprim directly stabilize the ternary complex. If the indirect mechanism is responsible, the interaction energy between the protein and trimethoprim would be expected to improve in the presence of NADPH. The calculated intermolecular energy between trimethoprim and the protein in the binary complex was -214.4 kcal mol-1 compared with -188.7 kcal mol-1 in the presence of NADPH. The major difference in binding energy arose because His45 becomes protonated (Poe et al. 1979) upon binding of NADPH, as indicated by proton magnetic resonance studies.
The interaction of this protonated histidine with the positively charged trimethoprim accounts for 20.9 kcal mol-1 of the 25.7 kcal mol-1 difference in the interaction energy. (Note the importance of correctly identifying the charge state of His45.)
Thus, the energy results indicate that NADPH binding does not predispose the enzyme to interact more favorably with trimethoprim, and protein-ligand interactions cannot explain the observed cooperativity in E. coli DHFR.
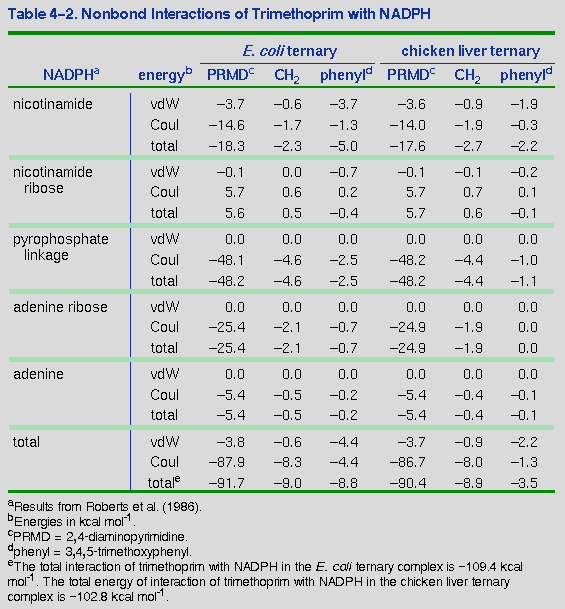
Comparison of the interactions of NADPH with trimethoprim in the E. coli ternary complex and the chicken liver ternary complex provides a clue as to the source of the enhanced cooperativity of the bacterial enzyme with NADPH. The energy of this interaction in the minimized structures is 6.6 kcal mol-1 more favorable in the E. coli complex. This is not sufficient to overcome the 25.7 kcal mol-1 increase in energy experienced upon binding of NADPH and indicates that quantitative energy comparisons may be unreliable in this model. Nevertheless, the qualitative trend is still worth investigating more closely. By partitioning these interaction energies into contributions from the various functional groups, the specific interactions responsible for this difference can be determined (Table 4-2). The favorable interactions arise predominantly from the spatial relationship between the trimethoxyphenyl ring of trimethoprim and NADPH. The trimethoxyphenyl ring occupies a different cleft in the chicken liver enzyme, so that the ring is closer to the nicotinamide of NADPH and its associated ribose in the E. coli system than in the chicken liver ternary complex. Table 4-2 shows that the van der Waals interactions between these groups of the NADPH and the trimethoprim phenyl ring are 2.2 kcal mol-1 more favorable in the E. coli complex. Coulombic interactions between the entire NADPH molecule and the trimethoxyphenyl ring are 3.1 kcal mol-1 more favorable in the E. coli ternary complex. This suggests that it is the difference in trimethoprim-NADPH interactions that underlies the cooperativity in the E. coli complex. Specifically, the cooperativity appears to be due to better van der Waals interactions between NADPH and the trimethoxyphenyl ring of trimethoprim in the E. coli complex than in the vertebrate DHFR.
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Minimization access
Minimization access
 Minimization - General Methodology
Minimization - General Methodology
 Minimization - Vibrational Calculation
Minimization - Vibrational Calculation
Copyright Biosym/MSI
{kind=link}
{kind=link}
{kind=link}