 Periodic Boundary
Conditions Periodic Boundary
Conditions
Periodic Boundary
Conditions Periodic Boundary
Conditions
The Discover program accepts only Cartesian coordinates. This is important when using asymmetric space groups, since the symmetry operators assume that the input coordinates correspond to the standard asymmetric unit as defined in the International Tables for Crystallography (Reidl 1983).
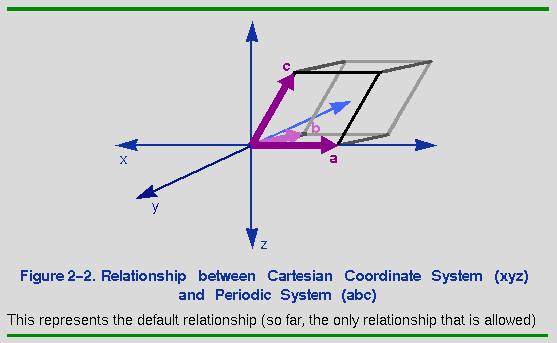
It is assumed that the x Cartesian axis corresponds to the a crystal axis and that the b axis lies in the x,y plane (Figure 2-2).
All the molecules in an asymmetric unit or unit cell must be complete molecules for calculations with Discover version 2.9.5, since it cannot handle bonds between symmetrically related objects. However, Discover version 95.0 can handle bonds across cell boundaries.
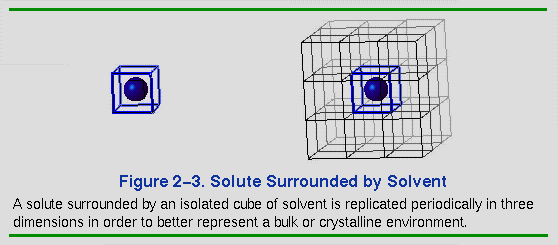
The left side of Figure 2-3 shows a solute molecule surrounded by enough solvent to occupy the volume (and shape) of a cube. A simulation carried out on this isolated cubic system is a poor approximation of what would happen in a true bulk solvent environment. For example, the solute can diffuse toward a surface or solvent molecules can evaporate. To remedy this, on the right of Figure 2-3 the cube is replicated in three dimensions to form a 3 x 3 x 3 lattice of identical cubes. This is a much better representation of bulk solvent for the interior cube, because molecules near the surfaces now interact with solvent in adjacent cubes. The imaged atoms are used to calculate energies and forces on the real atoms in the interior cube. The energies and forces on the imaged atoms themselves are not calculated because their motions are computed as symmetry operations on the real atoms, for example, by translations along the cubic axes.
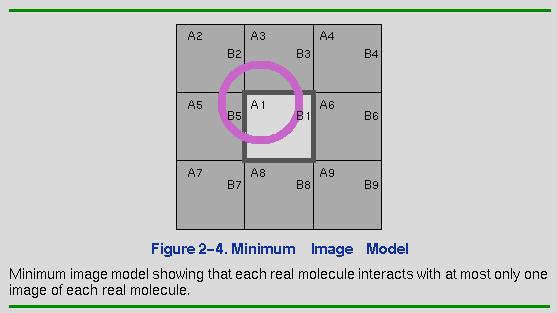
Consider the implications of this model for a specific case. In Figure 2-4, molecule A1 is located near an edge of the square. (For simplicity, this discussion focuses on a two-dimensional lattice.) In addition, eight images of A1 (A2-A9) are present in the adjacent symmetrically related squares. Consider the interactions of molecules A with molecules B. The closest image of B to A1 is actually not B1, but rather B5. If molecules in the interior cell are allowed to interact only with the molecule or molecular image closest to it, this is called a minimum-image model. Each molecule interacts only with those molecules and images within a distance of half the cell size. The advantage of this approach is its simplicity. It is straightforward to compute as required between a given pair of molecules without explicitly keeping track of the images in neighboring cells. All periodic boundary algorithms imply a cutoff criterion, but the minimum-image convention implies a maximum distance for this cutoff of no more than half the cell dimensions.
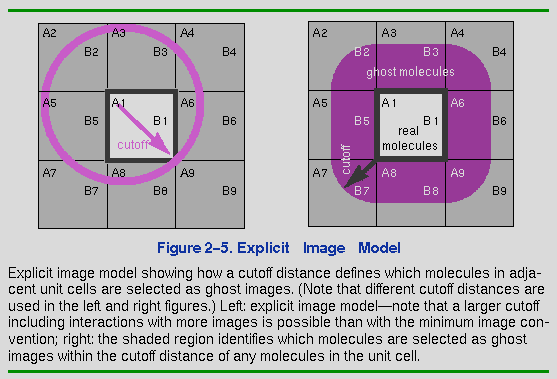
Ghost molecules shadow their symmetrically related counterparts. For example, the left side of Figure 2-5 shows molecule A1 interacting with several images of B (B1, B2, B3, B5) within the specified cutoff radius (shown as a magenta circle centered on A1). Note, in fact, that A1 interacts with several of its own images as well (A3, A5, A6, A8). However, when it comes time to move the molecules (in a dynamics step or minimization iteration), only the real molecules (A1 and B1) are moved according to the accumulated forces each molecule has felt. The ghost molecule positions are simply regenerated by applying the defined symmetry relations to the new positions of the molecules.
The right side of Figure 2-5 shows which molecules in the adjacent unit cells become explicit ghost molecules for a given cutoff distance. Note that not every molecule in an adjacent cell becomes a ghost. However, if a cutoff distance that is longer than the cell length is used, ghosts from unit cells beyond the nearest neighbor cells may be included. As molecules move in and out of the boundaries, the molecules that are ghosts can change. Therefore, the ghost list is regenerated periodically. Note that nonbond interactions do not have to be calculated between ghost atoms. This helps to significantly reduce computational time.
 Main
access page
Main
access page  Theory/Methodology access.
Theory/Methodology access.
 Describe-System access
Describe-System access
 Empirical Fit of the Surface
Empirical Fit of the Surface
 Nonbond Cutoffs
Nonbond Cutoffs
Copyright Biosym/MSI
{kind=link}
{kind=link}
{kind=link}
{kind=link}