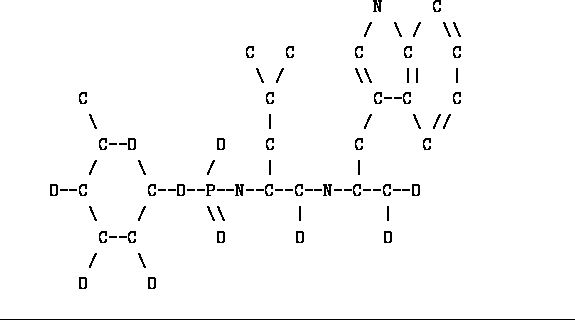
Figure: Unprocessed Phosphoramidon
Even though most of the geometry restraints you will ever need are supplied with TNT you may encounter the need to define the restraints for a group yourself. If you are working with enzyme-inhibitor complexes you will certainly have to. However, even in the case of a simple protein you may encounter the binding of an unusual solvent molecule or discover some strange chemistry.
The definition of stereochemistry restraints requires that you analyze the molecule to determine which parts can be described using the given libraries. Then restraints for the unique parts must be be created. These steps are illustrated with the following example.
Figure ![]() shows the structure of the
phosphoramidon inhibitor of Thermolysin. This is a natural product which
is a reasonably good inhibitor.
We will step through the process of defining the standard
geometry of this rather strange compound.
shows the structure of the
phosphoramidon inhibitor of Thermolysin. This is a natural product which
is a reasonably good inhibitor.
We will step through the process of defining the standard
geometry of this rather strange compound.
The first step when defining a new structure or group is to see if one can break it into smaller, known groups. (Because we often deal with proteins, the natural units of structure are referred to as ``residues'', but it should be understood that a ``residue'' can be any collection of atoms that the heart desires.) Examining the above structure one will note that the atoms following the phosphorus (on the right) make up a leucine and a tryptophan linked by a normal peptide bond and ending in a normal C-terminus as found in proteins. Because these groups are already in the standard geometry library we will split each of them into separate residues. In proteins the C-terminus is handled by putting the extra oxygen in a separate residue, whose type is unimportant because it contains no restraints. The type of the link between the last amino acid and the residue containing the final oxygen is called CTERM. The names of these three residues -- the leucine, the tryptophan, and the terminal oxygen -- can be anything and I choose PEP1, PEP2, and MYEND.
The group of atoms to the left can be split several different ways or left as a single residue. It seems reasonable to keep the rhamnose sugar in one unit but what should be done with the pseudo-phosphate? It is a bad idea to make groups that are less than three atoms wide because no geometry restraint may span more than two residues and if a group were two atoms wide it would not be possible to define the torsion angles that would stretch from the preceding residue through the little residue and into the following residue. Therefore I have chosen to place the phosphorus and its two oxygen atoms into the same residue with the rhamnose sugar. I have named this residue SUGAR and named its type RHAM. Its link to the leucine (PEP1) will be called PHOSLINK.
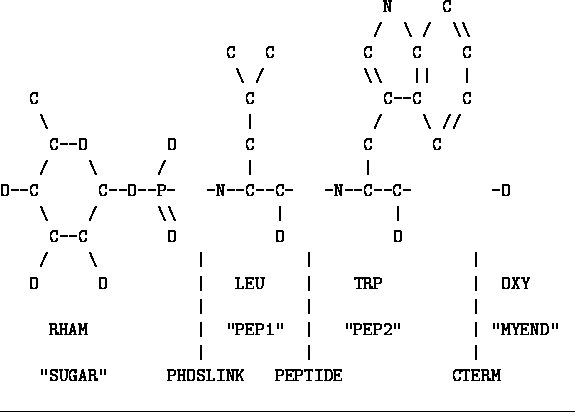
Figure: Phosphoramidon with residues and linkages named
In Figure ![]() I have redrawn the inhibitor after
splitting it in the manner I
discussed above and I have labeled the parts. The names
in quotes are the residue names and must be unique within the chain.
These names are included in the list of atomic coordinates (i.e. on the
ATOMC statements) and tell the program that a given atom belongs in the residue
that is specified.
The names under the vertical lines are the names of the linkage types that
connect the residues at these specified points. The other names
specify the type of each residue. These names are included on the
GEOMETRY statements that specify the geometry restraints of these groups.
I have redrawn the inhibitor after
splitting it in the manner I
discussed above and I have labeled the parts. The names
in quotes are the residue names and must be unique within the chain.
These names are included in the list of atomic coordinates (i.e. on the
ATOMC statements) and tell the program that a given atom belongs in the residue
that is specified.
The names under the vertical lines are the names of the linkage types that
connect the residues at these specified points. The other names
specify the type of each residue. These names are included on the
GEOMETRY statements that specify the geometry restraints of these groups.
With the structure now broken into residues and every part and link named we can write the RESIDUE statements that define to the program the type of each residue and the type of connection each residue makes.
Now that the residues of the inhibitor have been defined each atom must be given a name. The atom name must be unique within that residue but other residues in the structure may have atoms with the same name (e.g. CA). Because LEU, TRP, and CTERM are already defined in the standard geometry library for proteins, the atom names in those residues must conform to the same standard as the library. The standard geometry library uses the international convention for naming atoms within amino acids.
The residue type RHAM is not known to the library and we can choose any naming convention we wish. I choose to name the sugar ring's atoms in the normal fashion of numbering the atoms in the carbon chain starting from the most oxidized atom. Each oxygen was given the same number as its carbon. The phosphorus atom can easily be called P. The two oxygen atoms attached to P cannot be called O1 and O2 because those names are in use so I decided to call them PO1 and PO2. These are not the greatest names because some programs (but none in this package) determine the atom type by looking at the first letter of the atom name and these atoms would be improperly identified as phosphorus.
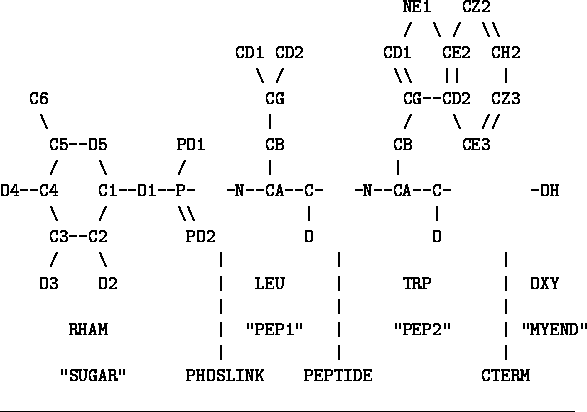
Figure: Phosphoramidon with atom names
In Figure ![]() I show the inhibitor with all the atoms
given their names. These names
must be used whenever an atom is referred to by the data given to a program.
These names will be on the ATOMC statements defining the coordinates of these
atoms and will also be used on the GEOMETRY statements that define the
restraints to be applied.
I show the inhibitor with all the atoms
given their names. These names
must be used whenever an atom is referred to by the data given to a program.
These names will be on the ATOMC statements defining the coordinates of these
atoms and will also be used on the GEOMETRY statements that define the
restraints to be applied.
Now we must look at the residue types and linkage types and determine which still need to be defined and which we are finished with. LEU, TRP, PEPTIDE, and CTERM are in the protein library and need not be worried about any more. OXY contains a single atom and does not need any restraints. Therefore it does not need to be worried about either. All that need be defined are RHAM and PHOSLINK.
Here we must come to grips with the difference between a residue type and a linkage type. A residue type contains the definitions of a collection of geometry restraints that involve only atoms within a single residue. A linkage type contains the definitions of restraints that span two residues but may also contain restraints that involve only atoms within the first residue of the link or only atoms within the second residue. Therefore restraints must be defined in the linkage type if they span two residues, but many restraints can be placed in either the residue type or the linkage type. (If a restraint was placed in both it would end up with twice the weight). The decision as to where to specify the restraint is arbitrary, but some rules of convenience can be formulated.
When defining the geometry of a polymer composed of different kinds of monomers some restraints are common to all monomer types (e.g. within the backbone) and some restraints are unique to their particular type of monomer (e.g. the side chains). A good rule is to place only the unique restraints in the residue type and to place the general restraints in the linkage type, which is the same for all monomers. Consider a protein: each amino acid type has a CA to C bond restraint that is independent of the type of amino acid while the NE1 to CE2 bond restraint is unique to tryptophan. In these examples the NE1 to CE2 bond would be defined in the TRP residue type, while the CA to C bond would be defined in the PEPTIDE linkage type even though both atoms are in the same residue. If this rule were not followed and the CA to C bond was defined in each amino acid type everything would work fine but this bond length would have to be defined separately for each amino acid, twenty different times.
Unfortunately this rule does not help us much with our present problem. We do not have a polymer composed of rhamnose sugars and strange phosphates and cannot define what would be a general linkage in such a polymer. However, rhamnose might come up in some future structure determination (It is a common carbohydrate) and it might be nice to have its standard geometry ready should the need arise. Therefore I decided to place geometry restraints that deal only with the rhamnose sugar into the RHAM residue type and to place all the restraints dealing with the phosphate and the link to PEP1 into PHOSLINK.
With my plan in mind I went in search of a small molecule structure determination of a rhamnose sugar molecule. My major source of such information is a book called ``Tables of Interatomic Distances and Configurations in Molecules and Ions'' published by the Chemical Society of London. (Of course, the Cambridge Structural Database is ideal for this task if you have ready access to it.) After leafing through many pages I found the structure I desired and wrote the geometry definition below. (Obviously one must ensure that the atom names are consistent with those used on the ATOMC statements.)
GEOMETRY RHAM BOND 1.42 0.02 C1, O5 GEOMETRY RHAM BOND 1.37 0.02 C1, O1 GEOMETRY RHAM BOND 1.51 0.02 C1, C2 GEOMETRY RHAM BOND 1.47 0.02 C2, O2 GEOMETRY RHAM BOND 1.53 0.02 C2, C3 GEOMETRY RHAM BOND 1.41 0.02 C3, O3 GEOMETRY RHAM BOND 1.52 0.02 C3, C4 GEOMETRY RHAM BOND 1.40 0.02 C4, O4 GEOMETRY RHAM BOND 1.56 0.02 C4, C5 GEOMETRY RHAM BOND 1.45 0.02 C5, O5 GEOMETRY RHAM BOND 1.52 0.02 C5, C6 GEOMETRY RHAM ANGLE 109 3 O5, C5, C4 GEOMETRY RHAM ANGLE 110 3 O5, C5, C6 GEOMETRY RHAM ANGLE 114 3 C6, C5, C4 GEOMETRY RHAM ANGLE 109 3 C5, C4, C3 GEOMETRY RHAM ANGLE 110 3 C5, C4, O4 GEOMETRY RHAM ANGLE 108 3 O4, C4, C3 GEOMETRY RHAM ANGLE 111 3 C4, C3, C2 GEOMETRY RHAM ANGLE 114 3 C4, C3, O3 GEOMETRY RHAM ANGLE 106 3 O3, C3, C2 GEOMETRY RHAM ANGLE 110 3 C3, C2, C1 GEOMETRY RHAM ANGLE 108 3 C3, C2, O2 GEOMETRY RHAM ANGLE 104 3 O2, C2, C1 GEOMETRY RHAM ANGLE 113 3 C2, C1, O5 GEOMETRY RHAM ANGLE 109 3 C2, C1, O1 GEOMETRY RHAM ANGLE 109 3 O1, C1, O5 GEOMETRY RHAM ANGLE 120 3 C1, O5, C5
Often such a book will not list the standard deviations for each geometry restraint and this book is no exception. Here I have used the numbers that Lynn Ten Eyck started using which are 0.02Å for bond lengths and 3 degrees for bond angles. We also use 15 degrees for torsion angles and 0.02Å for both types of planarity. The book did not list the torsion angle values or the handedness of each chiral center. Therefore I looked at the structure and guessed that the torsion angles should all be three-valued and staggered. This guess seems correct to me but I am not greatly concerned because I don't normally refine torsion angles and could choose not to define them at all. I also examined the structure of the molecule and wrote the definitions of the chiral centers. This completes the definition of RHAM.
GEOMETRY RHAM TORSION 3060 15 O5, C5, C4, C3 GEOMETRY RHAM TORSION 3060 15 C6, C5, C4, C3 GEOMETRY RHAM TORSION 3060 15 C5, C4, C3, C2 GEOMETRY RHAM TORSION 3060 15 C4, C3, C2, C1 GEOMETRY RHAM TORSION 3060 15 C3, C2, C1, O5 GEOMETRY RHAM TORSION 3060 15 C2, C1, O5, C5 GEOMETRY RHAM CHIRAL 1 1 C1, O5, O1, C2 GEOMETRY RHAM CHIRAL 1 1 C2, O2, C1, C3 GEOMETRY RHAM CHIRAL 1 1 C3, O3, C2, C4 GEOMETRY RHAM CHIRAL 1 1 C4, O4, C5, C3 GEOMETRY RHAM CHIRAL 1 1 C5, O5, C6, C4
Now we arrive at the difficult task of defining PHOSLINK. (Difficult
not because of the program but because of not knowing what numbers to
plug in.) I checked the CRC and found that it said that
P-N bond lengths are 1.4910Å but had no mention of what structure that
number came from or what the bond angles should be. I looked in the
International Tables (Volume III) and found a table that included
P ![]() N
N ![]() (CH
(CH ![]() )
) ![]() (P-N of 1.60(0.03)Å) and
HPO
(P-N of 1.60(0.03)Å) and
HPO ![]() NH
NH ![]() (P-N of 1.78(0.06)Å).
I could not figure out the Lewis dot structure for the first one and
it didn't have any oxygen in it so I ignored it. I found HPO
(P-N of 1.78(0.06)Å).
I could not figure out the Lewis dot structure for the first one and
it didn't have any oxygen in it so I ignored it. I found HPO ![]() .NH
.NH ![]() in
the big book in the library and used the geometry from there to define
PHOSLINK. This is certainly not the best structural analog because
the atoms bonded to the phosphorus have no carbon atoms bonded to them.
All of the P-O bond lengths did not seem to vary much between different
structures so I defined those bond lengths with the normal sigma. I gave
the P-N bond length a larger sigma because I was quite uncertain about
its real value. It is interesting to note that during refinement the
P-N bond length shortened to about 1.5Å. This possibly implies that
incorrect geometry definitions can be overcome if the crystallographic
data are good enough.
in
the big book in the library and used the geometry from there to define
PHOSLINK. This is certainly not the best structural analog because
the atoms bonded to the phosphorus have no carbon atoms bonded to them.
All of the P-O bond lengths did not seem to vary much between different
structures so I defined those bond lengths with the normal sigma. I gave
the P-N bond length a larger sigma because I was quite uncertain about
its real value. It is interesting to note that during refinement the
P-N bond length shortened to about 1.5Å. This possibly implies that
incorrect geometry definitions can be overcome if the crystallographic
data are good enough.
GEOMETRY PHOSLINK BOND 1.52 0.02 -O1, P GEOMETRY PHOSLINK BOND 1.52 0.02 P, PO1 GEOMETRY PHOSLINK BOND 1.52 0.02 P, PO2 GEOMETRY PHOSLINK BOND 1.78 0.06 P, +N GEOMETRY PHOSLINK ANGLE 115 3 -O1, P, PO1 GEOMETRY PHOSLINK ANGLE 115 3 -O1, P, PO2 GEOMETRY PHOSLINK ANGLE 115 3 PO1, P, PO2 GEOMETRY PHOSLINK ANGLE 103 3 -O1, P, +N GEOMETRY PHOSLINK ANGLE 103 3 PO1, P, +N
This is the definition of the geometry restraints in PHOSLINK. I did not define torsion angles because I was not sure what they should be and I wasn't going to refine them anyway. Note that some atom names have a ``-'' in front of them and some do not. The program treats both classes the same and looks for those atoms in the first residue in the linked pair (SUGAR). The atom names beginning with a ``+'' are in the second residue, PEP1.