


Next: Preparing for ARP/wARP
Up: Using ARP/wARP
Previous: Limitations
ARP/wARP is a command file driven package. These command files, although simple
in principle, turned out to be rather error prone. Many problems of previous
releases were due to these kind of errors. This distribution has been complemented
with a set of easy-to-use set-up scripts that we hope will help avoid mistakes
and will generally improve the overall user-friendliness of the programs.
THE USER IS STRONGLY ADVISED TO RUN ARP/WARP USING THE AUTOMATED SCRIPTS
PROVIDED WITH THE PACKAGE.
Of course, you may still generate your own
command files - see the section on 'I hate automated scripts'.
In fact, for the moment this might be a better choice for solvent building,
especially towards the end of refinement where you might want to keep very good control
of REFMAC parameters adjusting geometry weights, etc. In the next version we
hope to include a graphical interface giving you full access to more REFMAC parameters
rather than the basic set you can control now from the arp_warp_setup.sh script and
the warp.par parameters file.
The current automated scripts are:
- arp_warp_setup.sh This is the basic script for setting everything
up. Answering simple questions like which MTZ file, which labels to use, how many models,
how many processors, etc. allow a general parameter file, warp.par, to be created
which will be used for all subsequent jobs.
- arp_molrep.sh This is the script to run if you start from a reasonable
Molecular Replacement solution (application #1). This script will use the model
for providing geometrical restraints to the refinement. If your model is really bad,
but you have data extending to at least 2.0 Å resolution, we suggest to use the
warpNtrace.sh script (see below), which will auto-build a new model and will
keep on auto-building, thus providing a significantly higher radius of convergence.
- warp.sh This is the script
for applications #2 and #3. It allows to distribute jobs over a network, a
multi-processor or a stand-alone machine. It also performs multiple model
averaging if it is requested during setup. The script does not
require any external programs (other than CCP4).
- warpNtrace.sh This script is for auto-tracing and model building
(application #4). In some cases it can be considered to be used
also for application #1, instead of arp_molrep.sh.
Please note that this is an
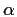 -version and only
SGI executables are provided at present. In brief, the script reads either a map
obtained after a job of warp.sh or a free-atoms model and does iterative
auto-tracing and refinement. It can practically deliver a complete auto-trace
of the model even with 2.0 Å data.
-version and only
SGI executables are provided at present. In brief, the script reads either a map
obtained after a job of warp.sh or a free-atoms model and does iterative
auto-tracing and refinement. It can practically deliver a complete auto-trace
of the model even with 2.0 Å data.
- side_dock.sh Takes a (partial) model auto-traced with warpNtrace.sh
and docks the fragments into the protein sequence.
- arp_solvent.sh The good old solvent building is now automated to even
greater extent. If you need to have a high degree of customisation of the REFMAC
part of the job, we recommend to run the old-style command files, at least
for the moment. Also, if you prefer to use SHELXL while water building,
you would again have to use the old-style files, as provided in the examples/ directory.
See the section 'I hate automated scripts' and the chapter on keywords.
Next: Preparing for ARP/wARP
Up: Using ARP/wARP
Previous: Limitations
VL AP RM
1998-09-03