The function minimize by this module, when the noncrystallographic symmetry is used as restraints is
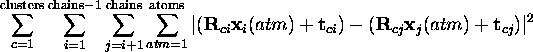
Thus, for each cluster there is a loop over all unique pairs of chains. The distance between each corresponding atom, after being moved to the prototypical location, is squared and summed.
A basic assumption of this module is that the objects which are related by the noncrystallographic symmetry are identified as individual chains, where the equivalent residues are given the same name in each chain. The old style of simply giving the second molecule's residues numbers much larger than the first will not work here. This assumption also means that the related molecules must be chemically identical. You cannot define noncrystallographic relations between two chains which are only similar in sequence, they must be identical.
When refining a model in the presence of noncrystallographic symmetry constraints the parameters of the model can be stored in the coordinate file in two states. In one state the model is expanded by the symmetry and the full asymmetric unit is present. In the other state only the unique atoms of the model are present. The nomenclature for these two states are the ``scattered" state and the ``gathered" state. The scatter and gather commands are used to change from one state to the other.
In the scattered state the atoms are named in the usual fashion. On the other hand, in the gathered state the atoms related by ncs symmetry are placed in a pseudo-chain given the name from the relevant CLUSTER statement. Because of this change in chain name a gathered coordinate set cannot be used in crystallographic calculations without being scattered first.